

Case Report
Unlocking the Mystery: A Rare Case of Ogilvie's Syndrome in Sickle Cell Disease
1 Post Graduate Trainee, Department of Medicine, Assam Medical College, Dibrugarh, Assam, India.
2 Senior Resident, Department of Medicine, Assam Medical College, Dibrugarh, Assam, India.
3 Professor and Head of Department, Department of Medicine, Assam Medical College, Dibrugarh, Assam, India.
*Corresponding Author: Aritra Saha, Senior Resident, Department of Medicine, Assam Medical College, Dibrugarh, Assam, India.
Citation: Duseja Y, Thakuria M, Saha A, Ajit K Pegu. (2024). Unlocking the Mystery: A Rare Case of Ogilvie's Syndrome in Sickle Cell Disease, Journal of Clinical Research and Clinical Trials, BioRes Scientia publishers. 3(1):1-5. DOI: 10.59657/2837-7184.brs.24.022
Copyright: © 2024 Aritra Saha, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: December 26, 2023 | Accepted: January 10, 2024 | Published: January 15, 2024
Abstract
Sickle cell disease (SCD) is an inherited haemoglobin disorder distinguished by reduced haemoglobin solubility, resulting in haemoglobin precipitation. This, in turn, leads to haemolysis, vaso-occlusion, and multiple organ dysfunction. While it is infrequent, sickle cell disease can occasionally manifest as intestinal obstruction. In this case report, we describe the presentation of a 13-year-old boy with sickle cell disease who exhibited symptoms of an acute abdomen. Ultimately, his condition was identified as pseudo-colonic obstruction, commonly referred to as Ogilvie syndrome, which is an exceedingly rare and seldom-reported complication of SCD.
Keywords: sickle cell disease; hemolytic anemia; ogilvie syndrome
Introduction
Acute colonic pseudo obstruction is also known as Ogilvie’s syndrome after Sir William H Ogilvie , in 1948 , who observed and reported this condition [1]. It is characterized by symptoms , signs , radiological pattern of large bowel obstruction with no evidence of mechanical obstruction [2]. Sickle cell disease is an inherited hemoglobinopathy where an unstable red blood cell becomes sickle-shaped under stress, many red blood cells polymerize, and the result is obstruction of blood vessels, which leads to complications such as Ogilvie’ s Syndrome [3]. An extremely less well known and potentially misinterpreted abdominal manifestation of sickle cell disease (SCD) is an acute colonic pseudo obstruction, or Ogilvie's syndrome[4].Here, we present the case of 13 year old boy with Sickle cell disease presented with evidence of bowel obstruction who was later diagnosed with Ogilvie’ syndrome and was managed conservatively without any surgical intervention.
Case Presentation
A 13 years old child, with known case sickle cell disease resident of northern region of the state of Assam, presented to the Medicine Outpatient Department with complaints of pain abdomen, vomiting and constitutional feature such as arthalgia and fatigue for a period 3 days. Pain abdomen was not radiating to back and was not relieved by bending forward or any change in position. There was no diurnal variation of pain. No association of any fever, rash or hematuria with pain. He is resident of northern region of same state. Patient has not passed stool for more than 4 days. There was no history of opioid use for analgesic. Patient received one packet of PRBC 6 months prior to the onset. On general examination, the patient was alert, conscious, well oriented to time, place and person.
On clinical examination his pulse was 128 beats per minute, BP was 94/64 mm hg, oxygen saturation was 98% on room air, respiratory rate was 30 per minute, body weight was 28 kg and temperature was normal. His abdomen was, distended, tender with sluggish bowel sounds. Per-abdominal examination revealed massive splenomegaly with palpable liver. Remaining systemic findings were within normal limits. His laboratory investigations are mentioned in Table-1.
Table 1
| Hemoglobin | 7.0gm/dl |
| WBC | 7700/cm |
| RBC | 3.4million/cum |
| Total Bilirubin | 7.21mg/dl |
| Unconjugated Bilirubin | 2.80mg/dl |
| Direct Bilirubin | 4.41mg/dl |
| AST | 93U/L |
| ALT | 202U/L |
Patient was kept nil per orally and was started on intravenous fluid (Ringers Lactate), parenteral non-steroidal anti-inflammatory (Ketorolac).
On reassessment there was persistent joint pain, with increasing distension and shortness of breath secondary to abdominal distension. On examination his abdomen was distended, tympanic and tender without hyperactive bowel sounds. Abdominal ultrasound revealed excessive gases in gut. Subsequently patient also developed features pneumonia with an oxygen saturation of 78% (with pulse oximtery) and consolidation in the Chest X Ray. Patient was started on empirical antibiotic therapy (Piperacillin-Tazobactam & Levofloxacin) and was shifted to ICU for better care. Patient responded to oxygen therapy via facemask and for the acute abdominal part, decompression with Ryle’s tube was done stimulate bowel activity. A repeat USG whole abdomen showed dilation of bowel loops [Fig.1].
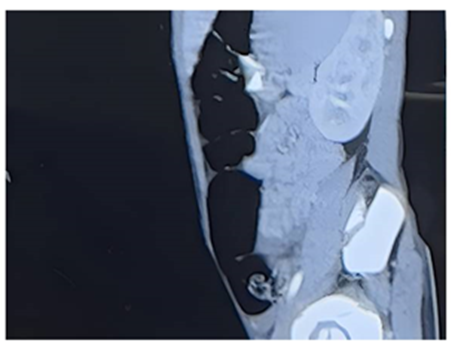
Figure 1: CECT Abdomen showing dilation of bowel loops.
CECT Abdomen was planned to rule out any obstructive cause of bowel dilation which revealed dilation of bowel loops upto proximal part of sigmoid colon with maximum diameter of 4.7cm [FIG.2] with enlarged spleen measuring approximately 16.4 cm.
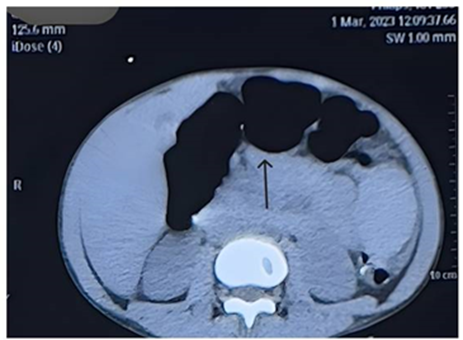
Figure 2: CECT Abdomen showing dilation of bowel loops.
The findings of the patient were suggestive of acute pseudo-colonic obstruction known as Ogilvie’s syndrome. Surgical opinion was obtained and a conservative approach with nasogastric decompression alone along with other supportive measures was advised. Patient responded to the treatment was successfully discharged after a period of 9 days. Patient is under follow up and has not experienced a fresh vaso-occlusive event subsequently.
Discussion
Sickle cell disease (SCD) refers to a group of hemoglobinopathies that include mutations in the gene encoding the beta subunit of hemoglobin[5].The sickle cell mutation occurs when negatively charged glutamine is replaced by a neutral valine at the sixth position of the beta-globin chain. The mutation is transmitted via Mendelian genetics and is inherited in an autosomal codominant fashion [6]. The most frequent and debilitating problem seen in patients with SCD are vaso-occlusive crises which often lead to severe organ dysfunction due to microvascular occlusion [7]. The incidence of these occlusive crises range between 0.8 and 1 episode per patient per year [7]. Abdominal pain and bowel hypotonia are commonest manifestations of an abdominal crisis. Although abdominal complications of SCD rarely involve the colon, there have been reports of ischemic colitis[8], necrotizing colitis [9] , nonspecific ulcerative colitis [10]. Pseudo-obstruction in the context of sickle cell disease is an uncommon occurrence. The predominant culprits behind secondary pseudo-obstruction are typically attributed to the dysfunction of various bodily systems, such as the gastrointestinal smooth muscle, nervous system, endocrine system, and immune system. Additionally, pseudo-obstruction may arise as a consequence of infections or surgical procedures [11].
The pathogenesis of pseudo obstruction, however, is uncertain and can be considered as diagnosis of exclusion[4]. Correction of the crisis also improves the pseudo obstruction, which suggests that the colonic symptoms are, indeed, a complication rather than coincidental [4]. The successful treatment of acute pseudo-obstruction with the parasympathomimetic drug such as neostigmine supports the theory that parasympathetic suppression may play a key role in the pathogenesis of the disease process [2,12,18]. Medical therapy for pseudo obstruction includes acetylcholinesterase inhibitors (neostigmine) to increase parasympathetic tone, prokinetics (erythromycin, cisapride, metoclopramide) to increase colonic motility, and analgesics for pain control [13]. One should aggressively treat the underlying medical condition, administer intravenous fluids, correct electrolyte abnormalities, make the patient nil per oral , and place a nasogastric tube and or a rectal tube for decompression [14]. The best evidence for medical treatment is available for neostigmine which is a reversible acetyl cholinesterase inhibitor so, indirectly stimulates the muscarinic receptors and increases the colonic motility [15].
Conclusion
Ogilvie’s syndrome is a rare manifestation in sickle cell disease in adolescent group and this case report introduces successful management of sickle cell induced Ogilvie’s with conservative management without any surgical intervention such as colonoscopic decompression or surgical exploration.
Declarations
Author Contribution
Conceptualization, Y.D. and A.S.; Methodology, Y.D. and M.T.: Validation, A.S. and A.K.P: Formal Analysis, Y.D.: Investigation, M.T. and Y.D.: Writing- original draft preparation, Y.D.: Writing- review and editing, M.T.: Supervision, A.S. and A.K.P.
Financial Support
No external funding received for this publication.
Conflict of Interest
Authors declare no conflict of interest associated with this publication.
References
- Vanek VW, Al-Salti M. (1986). Acute pseudo-obstruction of the colon (Ogilvie’s syndrome). An analysis of 400 cases. Dis Colon Rectum, 29:203-210.
Publisher | Google Scholor - De Giorgio R, Barbara G, Stanghellini V, et al. (2001). Review article: the pharmacological treatment of acute colonic pseudo-obstruction. Aliment Pharmacol Ther, 15:1717-1727.
Publisher | Google Scholor - Sedrak A, Kondamudi NP. (2023). Sickle Cell Disease. In: StatPearls. StatPearls Publishing: Treasure Island (FL).
Publisher | Google Scholor - Knox-Macaulay H, Ayyaril M, Nusrat N, Daar A. (2003). Colonic pseudo-obstruction in sickle cell disease. (Case Report). South Med J, 96:93-96.
Publisher | Google Scholor - Rees DC, Williams TN, Gladwin MT. (2010). Sickle-cell disease. Lancet Lond Engl, 376:2018-2031.
Publisher | Google Scholor - Steinberg MH, Sebastiani P. (2012). Genetic modifiers of sickle cell disease. Am J Hematol, 87:795-803.
Publisher | Google Scholor - Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. (1994). Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med, 330:1639-1644.
Publisher | Google Scholor - Karim A, Ahmed S, Rossoff LJ, Siddiqui R, Fuchs A, Multz AS. (2002). Fulminant ischaemic colitis with atypical clinical features complicating sickle cell disease. Postgrad Med J, 78:370-372.
Publisher | Google Scholor - Van der Neut FW, Statius van Eps LW, van Enk A, van de Sandt MM. (1993). Maternal death due to acute necrotizing colitis in homozygous sickle cell disease. Neth J Med, 42:132-133.
Publisher | Google Scholor - Terry SI, Rajendran A, Hanchard B, Serjeant GR. (1987). Ulcerative colitis in sickle cell disease. J Clin Gastroenterol, 9:55-57.
Publisher | Google Scholor - Dalgiç B, Sari S, Doğan I, Unal S. (2005). Chronic intestinal pseudoobstruction: report of four pediatric patients. Turk J Gastroenterol Off J Turk Soc Gastroenterol, 16:93-97.
Publisher | Google Scholor - Carcoforo P, Jorizzo EF, Maestroni U, Soliani G, Bergossi L, Pozza E. (2005). A new approach to the cure of the Ogilvie’s syndrome. Ann Ital Chir, 76:65-70.
Publisher | Google Scholor - Rj P, Md S, Mb K. (1999). Neostigmine for the treatment of acute colonic pseudo-obstruction. N Engl J Med, 341.
Publisher | Google Scholor - Khosla A, Ponsky TA. (2008). Acute colonic pseudoobstruction in a child with sickle cell disease treated with neostigmine. J Pediatr Surg, 43:2281-2284.
Publisher | Google Scholor - Saunders MD, Kimmey MB. (2004). Ogilvie’s syndrome. In: McDonald JWD, Burroughs AK, Feagan BG, editors. Evidence-based gastroenterology and hepatology.2nd ed. Oxford: Blackwell Publishing, 303-309.
Publisher | Google Scholor











