

Research Article
Sickle cells Diseased-Inherited RBC Disorders
1Department of chemistry, Sri JNMPG College Lucknow, U.P., India.
2Department of chemistry, Christ Church College Kanpur, U.P., India.
*Corresponding Author: D.K. Awasthi, Department of Chemistry, Sri J.N.M.PG College, Lucknow UP, India.
Citation: D.K. Awasthi, Kamal M. (2023). Sickle cells Diseased-Inherited RBC Disorders. Clinical and Laboratory Research, BioRes Scientia Publishers. 1(1):1-12. DOI: 10.59657/clr.brs.23.001
Copyright: © 2023 D.K. Awasthi, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: June 06, 2023 | Accepted: June 20, 2023 | Published: June 27, 2023
Abstract
Sickle cells Diseased is a group of inherited RBC disorders. The disease is named for the C-shaped farming tool known as a sickle. Sickle cells often become hard and sticky. This can increase the risk of blood clots. They also tend to die off early. This causes a constant shortage of RBCs an inherited disease in which the red blood cells have an abnormal crescent shape, block small blood vessels, and do not last as long as normal red blood cells. Sickle cell disease is caused by a mutation (change) in one of the genes for hemoglobin (the substance inside red blood cells that binds to oxygen and carries it from the lungs to the tissues). It is most common in people of West and Central African descent. It is also called sickle cell anemia.
Keywords: sickle cell; mutation; RBC; hemoglobin
Introduction
Sickle cell anemia, or sickle cell disease (SCD), is a genetic disease of the red blood cells (RBCs). Normally, RBCs are shaped like discs, which gives them the flexibility to travel through even the smallest blood vessels. However, with this disease, the RBCs have an abnormal crescent shape resembling a sickle. This makes them sticky and rigid and prone to getting trapped in small vessels, which blocks blood from reaching different parts of the body. This can cause pain and tissue damage.
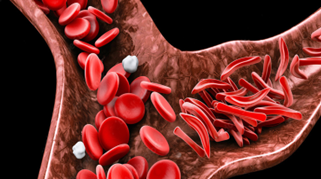
Figure 1: Sickle cell anemia
Symptoms of sickle cell anemia usually show up at a young age. They may appear in babies as early as 4 months old, but generally occur around the 6-month mark.
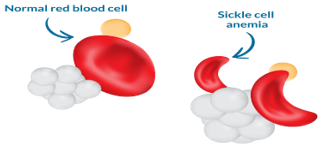
Figure 2
While there are multiple types of SCD, they all have similar symptoms, which vary in severity. These include:
- excessive fatigue or irritability, from anaemia
- fussiness, in babies
- bedwetting, from associated kidney problems
- jaundice, which is yellowing of the eyes and skin
- swelling and pain in hands and feet
- frequent infections
- pain in the chest, back, arms, or legs
Types of sickle cell disease
Hemoglobin is the protein in red blood cells that carries oxygen. It normally has two alpha chains and two beta chains. The four main types of sickle cell anemia are caused by different mutations in these genes.
Hemoglobin SS disease
Hemoglobin SS disease is the most common type of sickle cell disease. It occurs when you inherit copies of the hemoglobin S gene from both parents. This forms hemoglobin known as Hb SS. As the most severe form of SCD, individuals with this form also experience the worst symptoms at a higher rate.
Hemoglobin SC disease
Hemoglobin SC disease is the second most common type of sickle cell disease. It occurs when you inherit the Hb C gene from one parent and the Hb S gene from the other. Individuals with Hb SC have similar symptoms to individuals with Hb SS. However, the anemia is less severe.
Hemoglobin SB+ (beta) thalassemia
Hemoglobin SB+ (beta) thalassemia affects beta globin gene production. The size of the red blood cell is reduced because less beta protein is made. If inherited with the Hb S gene, you will have hemoglobin S beta thalassemia. Symptoms are not as severe.
Hemoglobin SB 0 (Beta-zero) thalassemia
Sickle beta-zero thalassemia is the fourth type of sickle cell disease. It also involves the beta globin gene. It has similar symptoms to Hb SS anemia. However, sometimes the symptoms of beta zero thalassemia are more severe. It is associated with a poorer prognosis.
Hemoglobin SD, hemoglobin SE, and hemoglobin SO
These types of sickle cell disease are rarer and usually don’t have severe symptoms.
Sickle cell trait
People who only inherit a mutated gene (hemoglobin S) from one parent are said to have sickle cell trait. They may have no symptoms or reduced symptoms.
Children are only at risk for sickle cell disease if both parents carry sickle cell trait. A blood test called a hemoglobin electrophoresis can also determine which type you might carry.
People from regions that have endemic malaria are more likely to be carriers. This includes people from:
- Africa
- India
- the Mediterranean
- Saudi Arabia
Complications of sickle cell anemia
SCD can cause severe complications, which appear when the sickle cells block vessels in different areas of the body. Painful or damaging blockages are called sickle cell crises. They can be caused by a variety of circumstances, including:
- illness
- changes in temperature
- stress
- poor hydration
- altitude
The following are types of complications that can result from sickle cell anemia.
Severe anemia
Anemia is a shortage of RBCs. Sickle cells are easily broken. This breaking apart of RBCs is called chronic hemolysis. RBCs generally live for about 120 days. Sickle cells live for a maximum of 10 to 20 days.
Hand-foot syndrome
Hand-foot syndrome occurs when sickle-shaped RBCs block blood vessels in the hands or feet. This causes the hands and feet to swell. It can also cause leg ulcers. Swollen hands and feet are often the first sign of sickle cell anemia in babies.
Splenic sequestration
Splenic sequestration is a blockage of the splenic vessels by sickle cells. It causes a sudden, painful enlargement of the spleen. The spleen may have to be removed due to complications of sickle cell disease in an operation known as a splenectomy. Some sickle cell patients will sustain enough damage to their spleen that it becomes shrunken and ceases to function at all. This is called auto splenectomy. Patients without a spleen are at higher risk for infections from bacteria such as Streptococcus, Hemophilus, and Salmonella species.
Delayed growth
Delayed growth often occurs in people with SCD. Children are generally shorter but regain their height by adulthood. Sexual maturation may also be delayed. This happens because sickle cell RBCs can’t supply enough oxygen and nutrients.
Neurological complications
Seizures, strokes, or even coma can result from sickle cell disease. They are caused by brain blockages. Immediate treatment should be sought.
Eye problems
Blindness is caused by blockages in the vessels supplying the eyes. This can damage the retina.
Skin ulcers
Skin ulcers in the legs can occur if small vessels there are blocked.
Heart disease and chest syndrome
Since SCD interferes with blood oxygen supply, it can also cause heart problems
Lung disease
Damage to the lungs over time related to decreased blood flow can result in high blood pressure in the lungs (pulmonary hypertension) and scarring of the lungs (pulmonary fibrosis). These problems can occur sooner in patients who have sickle chest syndrome. Lung damage makes it more difficult for the lungs to transfer oxygen into the blood, which can result in more frequent sickle cell crises.
Priapism
Priapism is a lingering, painful erection that can be seen in some men with sickle cell disease. This happens when the blood vessels in the penis are blocked. It can lead to impotence if left untreated.
Gallstones
Gallstones are one complication not caused by a vessel blockage. Instead, they are caused by the breakdown of RBCs. A byproduct of this breakdown is bilirubin. High levels of bilirubin can lead to gallstones. These are also called pigment stones.
Sickle chest syndrome
Sickle chest syndrome is a severe type of sickle cell crisis. It causes severe chest pain and is associated with symptoms such as cough, fever, sputum production, shortness of breath, and low blood oxygen levels. Abnormalities observed on chest X-rays can represent either pneumonia or death of lung tissue (pulmonary infarction). The long-term prognosis for patients who have had sickle chest syndrome is worse than for those who have not had it.
Diagnosis
All new-borns in the United States are screened for sickle cell disease. Prebirth testing looks for the sickle cell gene in your amniotic fluid.
In children and adults, one or more of the following procedures may also be used to diagnose sickle cell disease.
Detailed patient history
This condition often first appears as acute pain in the hands and feet. Patients may also have:
severe pain in the bones
anemia
painful enlargement of the spleen
growth problems
respiratory infections
ulcers of the legs
heart problems
Your doctor may want to test you for sickle cell anemia if you have any of the symptoms mentioned above.
Blood tests
Several blood tests can be used to look for SCD: Blood counts can reveal an abnormal Hb level in the range of 6 to 8 grams per deciliter. Blood films may show RBCs that appear as irregularly contracted cells. Sickle solubility tests look for the presence of Hb S.
Hb electrophoresis
Hb electrophoresis is always needed to confirm the diagnosis of sickle cell disease. It measures the different types of hemoglobin in the blood.
Genetic View of Sickel Cell
Genes usually come in pairs: one copy of a gene comes from each biological parent. This pair of genes is known as a genotype. Sickle cell disease is caused by inheriting two copies (one from each parent) of an altered HBB gene, which causes the production of an abnormal form of beta (β)-globin, such as hemoglobin s(Hbs) Scientists have identified hundreds of variations in the HBB gene that cause abnormal beta-globin to form and cause disease. As a result, there is more than one type of sickle cell, which depends on the specific combination of alterations of the HBB gene you inherit. These different forms are described as your sickle cell genotype.
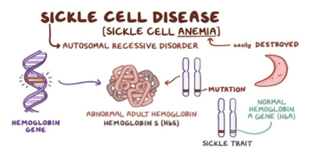
Figure 3: Sickle Cell Disease
The interactive diagram below, called a Punnett square, to see the likelihood of a child inheriting a form of sickle cell disease or sickle cell trait. This Punnett square can help you see how genes can be passed from a parent to a child in every single pregnancy, regardless of the genotypes of previous children. Note that the “sickle cell disease” selection for this tool is for the most common type of sickle cell, hemoglobin SS. However, you can create your own Punnett square by following the format below and using a different genotype (such as hemoglobin SC or hemoglobin S beta-zero) in place of “SS”.
Select a genotype (ie, AA, no sickle cell disease; AS, sickle cell trait carrier; or SS, sickle cell disease) for both the male and female, then select “View Results” to see the chances of a child inheriting sickle cell trait or sickle cell disease.
Normal HBB gene.
Sickle HBB gene.
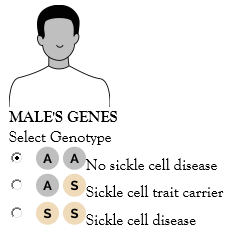
Figure 4
Bottom of Form
A person who carries the sickle cell trait inherits one copy of an abnormal (sickle) HBB gene and one copy of a normal HBB gene. This means that although their red blood cells contain some HbA, a portion of their red blood cells (20%-45%) consists of HbS. The levels of HbS in people with sickle cell trait are largely genetically determined. At rest, their red blood cells appear healthy—smooth and disc-shaped. However, under certain circumstances, their red blood cells can appear sickle shaped, and trait carriers can experience some of the Symptoms of sickle anemia. Some, but not all, genetic conditions can be inherited from one or both parents. Sickle cell anemia is one of these conditions. Its inheritance pattern is autosomal recessive. What do these terms mean? How exactly is sickle cell anemia passed on from parent to child? Geneticists use the terms dominant and recessive to describe the likelihood of a particular trait being passed on to the next generation. You have two copies of each of your genes one from your mother and another from your father. Each copy of a gene is called an allele. You may receive a dominant allele from each parent, a recessive allele from each parent, or one of each. Dominant alleles usually override recessive alleles, hence their name. For example, if you inherit a recessive allele from your father and a dominant one from your mother, you’ll usually display the trait associated with the dominant allele.
The sickle cell anemia trait is found on a recessive allele of the hemoglobin gene. This means that you must have two copies of the recessive allele — one from your mother and one from your father to have the condition. People who have one dominant and one recessive copy of the allele won’t have sickle cell anemia. Autosomal and sex-linked refer to the chromosome that the allele is present on. Each cell of your body typically contains 23 pairs of chromosomes. Out of each pair, one chromosome is inherited from your mother and the other from your father. The first 22 pairs of chromosomes are referred to as autosomes and are the same between males and females. The last pair of chromosomes are called sex chromosomes. These chromosomes differ between the sexes. If you’re female, you’ve received an X chromosome from your mother and an X chromosome from your father. If you’re male, you’ve received an X chromosome from your mother and a Y chromosome from your father. Some genetic conditions are sex-linked, meaning that the allele is present on the X or Y sex chromosome. Others are autosomal, meaning that the allele is present on one of the autosomes. The sickle cell anemia allele is autosomal, meaning it can be found on one of the other 22 pairs of chromosomes, but not on the X or Y chromosome. In order to have sickle cell anemia, you must have two copies of the recessive sickle cell allele. But what about those with only one copy? These people are known as carriers. They’re said to have sickle cell trait, but not sickle cell anemia. Carriers have one dominant allele and once recessive allele. Remember, the dominant allele usually overrides the recessive one, so carriers generally don’t have any symptoms of the condition. But they can still pass the recessive allele on to their children.
Here are a few example scenarios to illustrate how this might happen:
Scenario 1. Neither parent has the recessive sickle cell allele. None of their children will have sickle cell anemia or be carriers of the recessive allele.
Scenario 2. One parent is a carrier while the other isn’t. None of their children will have sickle cell anemia. But there’s a 50 percent chance that children will be carriers.
Scenario 3. Both parents are carriers. There’s a 25 percent chance that their children will receive two recessive alleles, causing sickle cell anemia. There’s also a 50 percent chance that they will be a carrier. Lastly, there’s also a 25 percent chance that their children won’t carry the allele at all.
Scenario 4. One parent isn’t a carrier, but the other has sickle cell anemia. None of their children will have sickle cell anemia, but they’ll all be carriers.
Scenario 5. One parent is a carrier and the other has sickle cell anemia. There’s a 50 percent chance that children will have sickle cell anemia and a 50 percent chance they’ll be carriers.
Scenario 6. Both parents have sickle cell anemia. All of their children will have sickle cell anemia.
If you have a family history of sickle cell anemia, but you don’t have it yourself, you may be a carrier. If you know others in your family, have it, or you’re not sure about your family history, a simple test can help to determine whether you carry the sickle cell allele. Sickle cell disease is a genetic red blood cell disorder. It changes normal, round red blood cells into cells shaped like crescent moons. Sickled cells can get stuck in blood vessels and block them, which stops oxygen from getting through. That can cause a lot of pain and can harm organs, muscles, and bones. Normal red blood cells move easily through blood vessels, taking oxygen to every part of your body. People who have sickle cell disease have mostly sickled red blood cells. These cells can get stuck and block blood flow through a blood vessel. That means red blood cells and oxygen can't flow to some parts of the body. Sickle cell anemia is a genetic condition that affects hemoglobin, the part of red blood cells responsible for carrying oxygen from the lungs to the rest of the body. Red blood cells with normal hemoglobin are round, smooth and flexible, allowing them to easily flow through blood vessels. In sickle cell anemia patients, the hemoglobin is abnormally shaped and sticks together, causing the red blood cells to become stiff and crescent, or sickle, shaped. These sickle-shaped red blood cells are more fragile and die more quickly than normally shaped red blood cells. Because the cells die faster than the body can create new ones, this creates a shortage of red blood cells.
Conditions such as high altitudes, severe dehydration, and low oxygen can lead to complications including:
Reduced blood supply to the spleen,
Muscle breakdown (rhabdomyolysis),
Kidney damage and chronic kidney disease,
Bleeding (hyphemia) and increased pressure in the eye (glaucoma) following eye injuries,
Sudden death with extreme exertion,
Kidney cancer (renal medullary carcinoma), exceedingly rare.
Because a person who carries the sickle cell trait may not experience symptoms, they may be unaware that they are a carrier. However, they are still able to pass the abnormal (sickle) HBB gene along to their children. A simple blood test from your doctor can determine if you are a carrier and at risk of passing along the abnormal gene. Sickle cell disease (SCD) is a serious inherited blood disorder where the red blood cells, which carry oxygen around the body, develop abnormally. The disorder mainly affects people of African, Caribbean, Middle Eastern, Eastern Mediterranean and Asian origin. In the UK, sickle cell disorders are most commonly seen in African and Caribbean people. Normal red blood cells are flexible and disc-shaped, but in SCD they can become rigid and shaped like a crescent (or sickle). The sickle-shaped cells contain defective hemoglobin (Hb), the iron-rich protein that enables red blood cells to carry oxygen from your lungs to the rest of the body. SCD is caused by a mutation in the hemoglobin, known as the ‘sickle’ mutation. The abnormal cells are unable to move around as easily as normal shaped cells and can block blood vessels, resulting in tissue and organ damage and episodes of life threatening and severe complications.
Such episodes are known as sickle cell crises or a Vaso-occlusive crises. They can last from a few minutes to several weeks. A sickle cell crisis is often described by a number of conditions. For example, it can cause an individual indescribable pain anywhere in their body, result in a cerebrovascular accident (CVA/stroke) and acute chest syndrome (ACS see below). The abnormal blood cells also have a shorter lifespan and aren't replaced as quickly as normal blood cells. This leads to a shortage of red blood cells, known as anemia. Symptoms of anemia include lethargy (a lack of energy), tiredness and breathlessness, particularly after exercise. Currently, treatment for SCD is limited and there is no cure except for bone marrow transplantation, which can be associated with many other risks. Experience with transplant in sickle cell is limited but specialists are becoming more skilled in the area and the number of bone marrow transplants undertaken is increasing. Treatment offered to individuals with SCD aims to be preventative and stop serious complications before they arise.
Causes sickle cell anemia
Sickle cell disease is caused by a mutation (an abnormal change) in the gene that instructs the body to produce hemoglobin. To have a diagnosis of a sickle cell disorder you will have to inherit the defective or mutated gene from both your mother and father. If you only inherit the gene from one parent, you are known as a sickle cell carrier, this is also referred to as sickle cell trait. It's likely that your blood will contain some sickle cells, but you will be able to produce normal hemoglobin and will not usually experience symptoms. In England, about 250,000 people are thought to have the sickle cell trait, with those of African-Caribbean origin primarily affected. As a carrier of SCD you may pass the gene on to any children you have. If you know you are a carrier of SCD it is important that you seek advice from your GP or are referred for genetic counselling before you and your partner conceive. This enables your partner to be screened for SCD and other blood disorders (such as Thalassemia) and informs you of the chances of your child inheriting SCD. If you are your partner are both affected by SCD there are services that you can be referred to in order to reduce the risk of conceiving a child with a sickle cell disorder. If two people with the sickle cell trait have a child, there's a one in four chance (25%) that the child will be born with sickle cell anemia. More information can be found at: The symptoms and process of sickle cell anemia can have a significant impact on a person's quality of life, both physically and psychologically. If complications develop, these can be very serious and potentially life threatening. There are numerous problems that someone with SCD can come up again; some individuals will experience numerous complications and some minimal difficulties throughout their life. Possible complications include (but are not limited to): Neurological damage. This can lead to cerebrovascular accidents (stroke) – where the blood supply to part of the brain is cut off. This can occur at any age and a high percentage of individuals with SCD will have had a stroke by the time they reach 18 years old. Regular monitoring is offered to children, which includes blood testing and Transcranial Dopplers (TCD) - which is similar to an ultrasound scan - looks at the rate of blood flow through the brain. Once any problems are detected treatment, such as blood exchanges are offered as a preventative measure. Once a person with SCD has had a stroke they are more likely to have further episodes. They will require close monitoring and ongoing treatment.
Increased vulnerability to infection. People with SCD do not have a functioning spleen and so are predisposed to infection. The spleen plays an important part in the ability to fight infection, as such individuals are recommended to take antibiotics for life in order to reduce the risk of certain infections. People with SCD are also encouraged to have an extended vaccination schedule. Although antibiotics are taken regularly, the risk of infection is still very high. Severe infections in someone with SCD are potentially fatal and often require hospital admission for intensive antibiotic treatment. Acute Chest Syndrome (ACS). In an ACS sickled red blood cells pool in the lung tissues. This can be precipitated by infection, fat embolism or decreased respiratory effort (often due to pain or following abdominal surgery). The pooling of sickle red blood cells in the lungs causes decreased air entry into the lungs and subsequently starves the body and its organs of oxygen. Emergency treatment is required, if left untreated an ACS can be fatal. Acute Chest Syndrome is the leading cause for death in adults with SCD; individuals who have had one ACS are more likely to have further episodes. Pulmonary Hypertension where the blood pressure inside the pulmonary arteries of the heart is raised. It causes the heart to become less efficient at moving the blood from the heart to the lungs. This decreases the amount of oxygen available to the muscles and around the body. It can cause shortness of breath and fatigue as well as many other associated problems. Once a person with SCD is found to have pulmonary hypertension, treatment is offered with the aim of slowing the progression of the disorder.
Acute and chronic pain. Pain is a common occurrence in SCD due to the blockages caused by the sickled red blood cells. Treatment involves a multimodal approach. This includes analgesia, psychological support, distraction techniques and complementary therapies. Pain can cause great distress and someone with SCD can experience pain every day. As such, it is important to support individuals and provide education on how best it can be managed. Many patients with SCD will manage their pain at home, however, at times people will need to be admitted to hospital and receive stronger and alternative treatments. After a flare up of pain or sickle cell crisis, it can take some time for an individual to recover completely and people often require a long period of recuperation. Leg ulcers are common in sickle cell disorders and have a high rate of reoccurrence. Leg ulcers can occur after injury or spontaneously. They can cause extreme pain and take months to heal. If an individual with SCD has a leg ulcer, once healed the skin at the site of the ulcer is extremely fragile and minimal trauma can cause a further breakdown. Treatment for leg ulcers can include frequent bandaging/ dressing, compression bandaging is sometimes necessary, bed rest, vitamin or mineral supplementation (zinc replacement). In severe cases blood transfusion or exchange can often aid the recovery process. Eye complications. Complications related to sickle cell can occur slowly over a period of time or be instant. People with SCD should have yearly eye checks to ensure any problems are identified and acted on as appropriate. Eye complications can result in visual changes/ reduced vision or complete loss of vision.
Sickle cell anemia is treated
A number of different treatments are available for SCD: Rehydration with intravenous fluids helps red blood cells return to a normal state. The red blood cells are more likely to deform and assume the sickle shape if you’re dehydration. Treating underlying or associated infections is an important part of managing the crisis, as the stress of an infection can result in a sickle cell crisis. An infection may also result as a complication of a crisis. Blood transfusions improve transport of oxygen and nutrients as needed. Packed red cells are removed from donated blood and given to patients. Supplemental oxygen is given through a mask. It makes breathing easier and improves oxygen levels in the blood. Pain medication is used to relieve the pain during a sickle crisis. You may need over-the-counter drugs or strong prescription pain medication like morphine. (Droxia, Hydria) helps to increase production of fetal hemoglobin. It may reduce the number of blood transfusions. Immunizations can help prevent infections. Patients tend to have lower immunity. Bone marrow transplant has been used to treat sickle cell anemia. Children younger than 16 years of age who have severe complications and have a matching donor are the best candidates.
Home care
There are things you can do at home to help your sickle cell symptoms: Use heating pads for pain relief, take folic acid supplements, as recommended by your doctor. Eat an adequate number of fruits, vegetables, and whole-wheat grains. Doing so can help your body make more RBCs. Drink more water to reduce the chances of sickle cell crises.
Exercise regularly and reduce stress to reduce crises, too. Contact your doctor immediately if you think you have any type of infection. Early treatment of an infection may prevent a full-blown crisis.
Sickle cell test
A sickle cell test is a simple blood test used to determine if you have sickle cell disease (SCD) or sickle cell trait. People with SCD have red blood cells (RBCs) that are abnormally shaped. Sickle cells are shaped like a crescent moon. Normal RBCs look like doughnuts. The sickle cell test is part of routine screening performed on a baby after they’re born. However, it can be used on older children and adults when needed.
Sickle cell trait
People with sickle cell trait are genetic carriers of SCD. They have no symptoms and can’t develop SCD, but they may be able to pass it on to their children. Those with the trait may have a higher risk of some other complications, including unexpected exercise-related death. New-born are regularly screened for SCD soon after birth. Early diagnosis is key. This is because children with SCD may be more vulnerable to serious infections within weeks of birth. Testing early helps ensure infants with SCD get the proper treatment to protect their health. No preparation is required for the sickle cell test. However, receiving a sickle cell test within 90 days after a blood transfusion may lead to inaccurate test results. Transfusion can reduce the amount of hemoglobin S the protein that causes SCD in the blood. A person who’s undergone a recent transfusion may have a normal sickle cell test result, even if they have SCD. The sickle cell test is a normal blood test. Complications are extremely rare. You may feel a little light headed or dizzy after the test, but these symptoms will go away when you sit down for a few minutes. Eating a snack may also help.
The puncture wound has a slim chance of becoming infected, but the alcohol swab used prior to the test normally prevents this. Apply a warm compress to the site if you develop abrusie. It is a genetic condition that’s present from birth. Many genetic conditions are caused by altered or mutated genes from your mother, father, or both parents.
People with sickle cell anemia have red blood cells that are shaped like a crescent or sickle. This unusual shape is due to a mutation in the hemoglobin gene. Hemoglobin is the molecule on red blood cells that allows them to deliver oxygen to tissues throughout your body. The sickle-shaped red blood cells can lead to a variety of complications. Due to their irregular shape, they can become stuck within blood vessels, leading to painful symptoms. Additionally, sickle cells die off faster than typical red blood cells, which can lead to anemia.
Some, but not all, genetic conditions can be inherited from one or both parents. Sickle cell anemia is one of these conditions. Its inheritance pattern is autosomal recessive. What do these terms mean? How exactly is sickle cell anemia passed on from parent to child? Geneticists use the terms dominant and recessive to describe the likelihood of a particular trait being passed on to the next generation. You have two copies of each of your genes one from your mother and another from your father. Each copy of a gene is called an allele. You may receive a dominant allele from each parent, a recessive allele from each parent, or one of each. Dominant alleles usually override recessive alleles, hence their name. For example, if you inherit a recessive allele from your father and a dominant one from your mother, you’ll usually display the trait associated with the dominant allele.
The sickle cell anemia trait is found on a recessive allele of the hemoglobin gene. This means that you must have two copies of the recessive allele — one from your mother and one from your father to have the condition. People who have one dominant and one recessive copy of the allele won’t have sickle cell anemia. Autosomal and sex-linked refer to the chromosome that the allele is present on. Each cell of your body typically contains 23 pairs of chromosomes. Out of each pair, one chromosome is inherited from your mother and the other from your father. The first 22 pairs of chromosomes are referred to as autosomes and are the same between males and females. The last pair of chromosomes are called sex chromosomes. These chromosomes differ between the sexes. If you’re female, you’ve received an X chromosome from your mother and an X chromosome from your father. If you’re male, you’ve received an X chromosome from your mother and a Y chromosome from your father. Some genetic conditions are sex-linked, meaning that the allele is present on the X or Y sex chromosome. Others are autosomal, meaning that the allele is present on one of the autosomes. The sickle cell anemia allele is autosomal, meaning it can be found on one of the other 22 pairs of chromosomes, but not on the X or Y chromosome. In order to have sickle cell anemia, you must have two copies of the recessive sickle cell allele. But what about those with only one copy? These people are known as carriers. They’re said to have sickle cell trait, but not sickle cell anemia. Carriers have one dominant allele and once recessive allele. Remember, the dominant allele usually overrides the recessive one, so carriers generally don’t have any symptoms of the condition. But they can still pass the recessive allele on to their children.
Here are a few example scenarios to illustrate how this might happen:
Scenario 1. Neither parent has the recessive sickle cell allele. None of their children will have sickle cell anemia or be carriers of the recessive allele.
Scenario 2. One parent is a carrier while the other isn’t. None of their children will have sickle cell anemia. But there’s a 50 percent chance that children will be carriers.
Scenario 3. Both parents are carriers. There’s a 25 percent chance that their children will receive two recessive alleles, causing sickle cell anemia. There’s also a 50 percent chance that they will be a carrier. Lastly, there’s also a 25 percent chance that their children won’t carry the allele at all.
Scenario 4. One parent isn’t a carrier, but the other has sickle cell anemia. None of their children will have sickle cell anemia, but they’ll all be carriers.
Scenario 5. One parent is a carrier and the other has sickle cell anemia. There’s a 50 percent chance that children will have sickle cell anemia and a 50 percent chance they’ll be carriers.
Scenario 6. Both parents have sickle cell anemia. All of their children will have sickle cell anemia.
If you have a family history of sickle cell anemia, but you don’t have it yourself, you may be a carrier. If you know others in your family, have it, or you’re not sure about your family history, a simple test can help to determine whether you carry the sickle cell allele. Sickle cell disease is a genetic red blood cell disorder. It changes normal, round red blood cells into cells shaped like crescent moons. Sickled cells can get stuck in blood vessels and block them, which stops oxygen from getting through. That can cause a lot of pain and can harm organs, muscles, and bones. Normal red blood cells move easily through blood vessels, taking oxygen to every part of your body. People who have sickle cell disease have mostly sickled red blood cells. These cells can get stuck and block blood flow through a blood vessel. That means red blood cells and oxygen can't flow to some parts of the body. Sickle cell anemia is a genetic condition that affects hemoglobin, the part of red blood cells responsible for carrying oxygen from the lungs to the rest of the body.
Red blood cells with normal hemoglobin are round, smooth and flexible, allowing them to easily flow through blood vessels. In sickle cell anemia patients, the hemoglobin is abnormally shaped and sticks together, causing the red blood cells to become stiff and crescent, or sickle, shaped. These sickle-shaped red blood cells are more fragile and die more quickly than normally shaped red blood cells. Because the cells die faster than the body can create new ones, this creates a shortage of red blood cells. The sickle-shaped cells also do not flow as easily through blood vessels. They are sticky and can catch on one another, causing the cells to pile up and block the blood vessel. This can cause tissue and organ damage, and sometimes stroke.
Treatment
The only cure for sickle cell anemia is a bone marrow transplant, also known as stem cell transplant. During this procedure the patient's own bone marrow is replaced by donor bone marrow cells, which produce new red blood cells for the body. This procedure can have serious side effects, and finding a matching donor can be challenging. Because of this, this treatment is not usually performed unless the patient is severely affected by sickle cell anemia. Otherwise, treatment will generally focus on managing the symptoms and maintaining the patient's quality of life.
Young children diagnosed with the condition are often given an antibiotic to prevent infections, such as pneumonia. Adults may also be given antibiotics to help fight off infections. Vaccinations, for adults and especially for young children, are also important to help prevent deadly infections. Over-the-counter pain medications or home remedies, such as applying heat to the affected area, can help manage the pain some patients experience. Sometimes stronger pain medication is needed, and prescribed by their medical team. Patients may also be given a medication to help the body create fetal hemoglobin, which is found in new-born and can prevent the formation of sickle-shaped cells. The long-term effects of this medication are still being researched, and parents should talk to their medical team to determine whether it is a good treatment plan for their child. Since sickle cells can stick together and cause blockages in the blood vessels, special attention should be paid to the risk of stroke.
Conclusion
People who have sickle cell disease have mostly sickled red blood cells. These cells can get stuck and block blood flow through a blood vessel. That means red blood cells and oxygen can't flow to some parts of the body. Sickle cell anemia is a genetic condition that affects hemoglobin, the part of red blood cells responsible for carrying oxygen from the lungs to the rest of the body. Red blood cells with normal hemoglobin are round, smooth and flexible, allowing them to easily flow through blood vessels. In sickle cell anemia patients, the hemoglobin is abnormally shaped and sticks together, causing the red blood cells to become stiff and crescent, or sickle, shaped.
References
- (2014). Evidence-Based Management of Sickle Cell Disease: Expert Panel Report.
Publisher | Google Scholor - Schultz CL, Tchume-Johnson T, Jackson T, Enninful-Eghan H, Schapira MM, Smith-Whitley K. (2020). Reproductive intentions in mothers of young children with sickle cell disease. Pediatr Blood Cancer, 67(5):e28227.
Publisher | Google Scholor - Boafor TK, Olayemi E, Galadanci N, et al. (2016). Pregnancy outcomes in women with sickle-cell disease in low and high income countries: a systematic review and meta-analysis.. BJOG, 123(5):691-698.
Publisher | Google Scholor - Justine Gellen-Dautremer, Sylvain Le Jeune, Marie-Catherine Receveur, Elena Foïs. (2019). Hydroxyurea Exposure throughout Pregnancy in Patients with Sickle-Cell Disease :4 Case Reports from European Non-Interventional, Multicentric, Prospective Escort-HU Study. Blood, 134(1):1027-1027.
Publisher | Google Scholor - Brandow AM, Carroll CP, Creary S, et al. (2020). American Society of Haematology 2020 guidelines for sickle cell disease: management of acute and chronic pain. Blood Adv. 4(12):2656-2701.
Publisher | Google Scholor - Howard J, Hart N, Roberts-Harewood M, et al. (2015). Guideline on the management of acute chest syndrome in sickle cell disease. Br J Haematol, 169(4):492-505.
Publisher | Google Scholor - Brandow AM, DeBaun MR. (2018). Key Components of Pain Management for Children and Adults with Sickle Cell Disease. Hematol Oncol Clin North Am, 32(3):535-550.
Publisher | Google Scholor - Lam T, Nagappa M, Wong J, Singh M, Wong D, Chung F. (2017). Continuous Pulse Oximetry and Capnography Monitoring for Postoperative Respiratory Depression and Adverse Events. Anesth Analg, 125(6):2019-2029.
Publisher | Google Scholor - Swegle JM, Logemann C. (2006). Management of common opioid-induced adverse effects. Am Fam Physician, 74(8):1347-1354.
Publisher | Google Scholor - Gong L, Parikh S, Rosenthal PJ, Greenhouse B. (2013). Biochemical and immunological mechanisms by which sickle cell trait protects against malaria. Malar J. 12(1):317.
Publisher | Google Scholor - Chou ST, Alsawas M, Fasano RM, et al. (2020). American Society of Haematology 2020 guidelines for sickle cell disease: transfusion support. Blood Adv, 4(2):327-355.
Publisher | Google Scholor - Davis BA, Allard S, Qureshi A, et al. Guidelines on red cell transfusion in sickle cell disease Part II: indications for transfusion. Br J Haematol., 176(2):192-209.
Publisher | Google Scholor - Hoppe C, Neumayr L. (2021). Sickle Cell Disease: Monitoring, Current Treatment, and Therapeutics Under Development. Hematol Oncol Clin North Am., 33(3):355-371.
Publisher | Google Scholor - (2019). Acute Chest Syndrome.
Publisher | Google Scholor - DeBaun MR, Jordan LC, King AA, et al. (2020). American Society of Haematology 2020 guidelines for sickle cell disease: prevention, diagnosis, and treatment of cerebrovascular disease in children and adults. Blood Advances., 4(8):1554-1588.
Publisher | Google Scholor - (2021). Sickle cell disease in children and adolescents: diagnosis, guidelines for comprehensive care, and care paths and protocols for management of acute and chronic complications.
Publisher | Google Scholor - Ochocinski D, Dalal M, Black LV, et al. (2020). Life-Threatening Infectious Complications in Sickle Cell Disease: A Concise Narrative Review. Frontiers in Pediatrics.
Publisher | Google Scholor - Ellison AM, Smith Whitley K, Kittick M, et al. (2018). A Standardized Clinical Pathway to Decrease Hospital Admissions Among Febrile Children with Sickle Cell Disease. J Pediatr Hematol Oncol., 40(2):111-115.
Publisher | Google Scholor - Gilbert, DN; Chambers, HF. (2020). Sanford Guide to Antimicrobial Therapy 2020. Antimicrobial Therapy, Inc.
Publisher | Google Scholor - Baskin MN, Goh XL, Heeney MM, Harper MB. (2013). Bacteraemia Risk and Outpatient Management of Febrile Patients with Sickle Cell Disease. Paediatrics, 131(6):1035-1041.
Publisher | Google Scholor - Rybak MJ, Lomaestro BM, Rotschafer JC, et al. (2009). Vancomycin Therapeutic Guidelines: A Summary of Consensus Recommendations from the Infectious Diseases Society of America, the American Society of Health‐System Pharmacists, and the Society of Infectious Diseases Pharmacists. Clin Infect Dis., 49(3):325-327.
Publisher | Google Scholor - Jordan LB, Smith-Whitley K, Treadwell MJ, Telfair J, Grant AM, Ohene-Frempong K. (2011). Screening U.S. College Athletes for Their Sickle Cell Disease Carrier Status. Am J Prev Med., 41(6):S406-S412.
Publisher | Google Scholor - Modell B, Darlison M. (2008). Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ.,86(6):480-487.
Publisher | Google Scholor - Salyer SW. (2007). Hematologic Emergencies. Elsevier, 555-574
Publisher | Google Scholor - Mitchell BL. (2018). Sickle Cell Trait and Sudden Death. Sports Med - Open. 4(1).
Publisher | Google Scholor - Quinn CT. (2013). Sickle cell disease in childhood: from newborn screening through transition to adult medical care. Pediatr Clin North Am., 60(6):1363-1381.
Publisher | Google Scholor - Darbari DS, Sheehan VA, Ballas SK. (2020). The vaso‐occlusive pain crisis in sickle cell disease: Definition, pathophysiology, and management. Eur J Haematol., 105(3):237-246.
Publisher | Google Scholor - Hernigou P, Daltro G, Flouzat-Lachaniette CH, Roussignol X, Poignard A. (2010). Septic arthritis in adults with sickle cell disease often is associated with osteomyelitis or osteonecrosis. Clin Orthop Relat Res, 468(6):1676-1681.
Publisher | Google Scholor - El-Haj N, Hoppe C. (2018). New-born Screening for SCD in the USA and Canada. Int J Neonatal Screen., 4(4):36.
Publisher | Google Scholor - US Preventive Services Task Force. (2008). Screening for sickle cell disease in newborns: recommendation statement. Am Fam Physician, 77(9):1300-1302.
Publisher | Google Scholor - (2008). ACMG ACT Sheets and Algorithms.
Publisher | Google Scholor - (2021). Newborn Screening ACT Sheet [FS] Sickle Cell Anaemia (HbSS Disease or HbS/Beta Zero Thalassemia).
Publisher | Google Scholor - Frömmel C. (2018). Newborn Screening for Sickle Cell Disease and Other Haemoglobinopathies: A Short Review on Classical Laboratory Methods—Isoelectric Focusing, HPLC, and Capillary Electrophoresis. Int J Neonatal Screen, 4(4):39.
Publisher | Google Scholor - (2012). ACMG ACT Sheets and Algorithms.
Publisher | Google Scholor - Caligiuri M, Levi MM, Kaushansky K, et al. (2015). Williams Hematology, 9E. McGraw-Hill Education / Medical.
Publisher | Google Scholor - Hollar MA. (2001). The Hair-on-End Sign. Radiology. 2001; 221(2):347-348.
Publisher | Google Scholor - Martí-Carvajal AJ, Solà I, Agreda-Pérez LH. (2019). Treatment for avascular necrosis of bone in people with sickle cell disease. Cochrane DatabaseSyst Rev. 12:CD004344.
Publisher | Google Scholor - Beckermann KE, Sharma D, Chaturvedi S, et al. (2017). Renal Medullary Carcinoma: Establishing Standards in Practice. J Oncol Pract., 13(7):414-421.
Publisher | Google Scholor - William BM, Corazza GR. (2007). Hyposplenism: A comprehensive review. Part I: Basic concepts and causes. Hematology. 12(1):1-13.
Publisher | Google Scholor - Shah R, Taborda C, Chawla S. (2017). Acute and chronic hepatobiliary manifestations of sickle cell disease: A review. World J Gastrointest Pathophysiol., 8(3):108-116.
Publisher | Google Scholor - Yawn BP, John-Sowah J. (2014). Management of Sickle Cell Disease: Recommendations from the 2014 Expert Panel Report. Am Fam Physician. 92(12):1069-1076.
Publisher | Google Scholor - Reeves SL, Tribble AC, Madden B, Freed GL, Dombkowski KJ. (2018). Antibiotic Prophylaxis for Children with Sickle Cell Anaemia. Pediatrics. 141(3).
Publisher | Google Scholor - Cober MP, Phelps SJ. (2015). Penicillin prophylaxis in children with sickle cell disease. J Pediatr Pharmacol Ther. 15(3):152-159.
Publisher | Google Scholor - Niihara Y, Miller ST, Kanter J, et al. (2018). A Phase 3 Trial of
l -Glutamine in Sickle Cell Disease. N Engl J Med. 2018; 379(3):226-235.
Publisher | Google Scholor - Vichinsky E, Hoppe CC, Ataga KI, et al. (2019). A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N Engl J Med.,381(6):509-519.
Publisher | Google Scholor - Ataga KI, Kutlar A, Kanter J, et al. (2017). Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N Engl J Med., 376(5):429-439.
Publisher | Google Scholor - Thom H, Jansen J, Shafrin J, et al. (2020). Crizanlizumab and comparators for adults with sickle cell disease: a systematic review and network meta-analysis. BMJ open. 2020; 10(9):e034147.
Publisher | Google Scholor - Kanter J, Liem RI, Bernaudin F, et al. (2021). American Society of Haematology 2021 guidelines for sickle cell disease: stem cell transplantation. Blood Adv, 5(18):3668-3689.
Publisher | Google Scholor - Galli S, Padayachee S, Howard J. (2013). The Use of Transcranial Doppler Ultrasonography in Adult Patients with Sickle Cell Disease: A Retrospective Analysis. Blood. 2013; 122(21):4692-4692.
Publisher | Google Scholor - Posovszky C, Wabitsch M. (2015). Regulation of Appetite, Satiation, and Body Weight by Enteroendocrine Cells. Part 1: Characteristics of Enteroendocrine Cells and Their Capability of Weight Regulation. Hormone Research in Paediatrics. 83(1):1-10.
Publisher | Google Scholor - Coates TD, Wood JC. (2017). How we manage iron overload in sickle cell patients. Br J Haematol., 177(5):703-716.
Publisher | Google Scholor - Ballas SK, Zeidan AM, Duong VH, DeVeaux M, Heeney MM. (2008). The effect of iron chelation therapy on overall survival in sickle cell disease and β-thalassemia: A systematic review. Am J Hematol, 93(7):943-952.
Publisher | Google Scholor - (2017). ACOG - Committee Opinion No. 692: Carrier Screening for Genetic Conditions.
Publisher | Google Scholor - Hustace T, Fleisher JM, Sanchez Varela AM, Podda A, Alvarez O. (2011). Increased prevalence of false positive haemoglobinopathy new-born screening in premature infants. Pediatr Blood Cancer, 57(6):1039-1043.
Publisher | Google Scholor - Longo D, Fauci A, Kasper D, Hauser S, Jameson J, Loscalzo J. (2011). Harrisons's Principles of Internal Medicine, 18th Edition, 2011. McGraw-Hill Medical.
Publisher | Google Scholor - Yawn et al. (2014). Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 312(10):1033-1048.
Publisher | Google Scholor - Al-Salem A. (2015). The Acute Chest Syndrome in Sickle Cell Anaemia. Springer.
Publisher | Google Scholor - Ballas SK, Lieff S, Benjamin LJ, et al. (2009). Definitions of the phenotypic manifestations of sickle cell disease. Am J Hematol.
Publisher | Google Scholor - Chaturvedi S, Ghafuri DL, Glassberg J, Kassim AA, Rodeghier M, et al. (2016). Rapidly progressive acute chest syndrome in individuals with sickle cell anaemia: a distinct acute chest syndrome phenotype. Am J Hematol. 91(12):1185-1190.
Publisher | Google Scholor - (2016). The management of sickle cell disease.
Publisher | Google Scholor - Dessap AM, Deux J-F, Abidi N, et al. (2011). Pulmonary Artery Thrombosis during Acute Chest Syndrome in Sickle Cell Disease. Am J Respir Crit Care Med, 184(9):1022-1029.
Publisher | Google Scholor - Vichinsky EP, Neumayr LD, Earles AN, et al. (2000). Causes and Outcomes of the Acute Chest Syndrome in Sickle Cell Disease. N Engl J Med, 342(25):1855-1865.
Publisher | Google Scholor - Otrock ZK, Thibodeaux SR, Jackups R. (2018). Vascular access for red blood cell exchange. Transfusion, 58 (S1):569-579.
Publisher | Google Scholor - Okomo U, Meremikwu MM. (2017). Fluid replacement therapy for acute episodes of pain in people with sickle cell disease. Cochrane Database of Systematic Reviews.
Publisher | Google Scholor













