

Case Report
Schnitzler Syndrome: Clinical Case and Review of The Literature
- N. Dimchova 1
- Broshtilova V 2*
- A. Batashki 3
- S. Bezhanova 1
- J. Velevska 4
- I. Yungareva 1
- S. Marina 1
1Medical Institute of the Ministry of Interior, Sofia, Bulgaria.
2Department of Dermatology and Venereology, Military Medical Academy, Sofia, Bulgaria.
3Department of Special Surgery, Medical University, Plovdiv, Bulgaria.
4Department of Infectious Diseases, Parasitology and Dermatovenereology, Medical University, Varna, Bulgaria.
*Corresponding Author: Broshtilova Valentina, Department of Dermatology and Venereology, Military Medical Academy, Sofia, Bulgaria.
Citation: Dimchova N., Broshtilova V., Batashki A., Bezhanova S., Velevska J., et al. (2023). Schnitzler Syndrome: Clinical Case and Review of The Literature. Dermatology Research and Reports. 2(1); DOI: 10.59657/2993-1118.brs.23.003
Copyright: © 2023 Broshtilova Valentina, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: May 10, 2023 | Accepted: May 20, 2023 | Published: May 30, 2023
Abstract
Schnitzler syndrome is a rare auto-inflammatory disease with a chronic relapsing course, characterized by recurrent urticarial lesions and monoclonal gammopathy. The manifestations of systemic inflammation are complemented by fever, bone and muscle pain, arthralgia/arthritis, lymphadenopathy, hepato- or splenomegaly, increased levels of acute inflammatory markers. A biological marker of the disease is the monoclonal gammopathy. Patients show high risk to develop AA-amyloidosis and lymphoproliferative diseases. A patient with a 5-year delay in diagnosis, who has responded well to systemic corticosteroids is presented. A comprehensive review of the new insights of disease pathogenesis and therapy is also highlighted.
Keywords: schnitzler syndrome; autoinflammatory disease; monoclonal gammopathy; interleukin-1
Introduction
Schnitzler syndrome (SS) was first described in 1972 as a chronic urticaria, accompanied by toxo-infectious syndrome [1]. To date, at about 300 cases have been reported worldwide [2]. The disease onset is usually in the 6 decades of age, rarely before 35 years, and with only one case aged 13 years [3, 4]. The syndrome reflects high male preponderance [2, 5].
SS is a rare autoinflammatory disease, characterized by urticarial rash and monoclonal gammopathy, usually of the IgM type, as a result of pathological dysregulation of interleukin-1 [2, 6-11]. Other less common findings are: recurrent fever, bone or joint pain, lymphadenopathy, headache, myalgia, arthralgia/arthritis, fatigue, weight loss, peripheral neuropathy, leukocytosis and/or elevated C-reactive protein (CRP) [4, 11-14]. The overall prognosis depends on progression to Waldenström macroglobulinemia, lymphomas, leukaemia, myeloma and other lymphoproliferative malignancies [4, 12, 13, 15-18]. Evolution to AA amyloidosis has also been described [4, 19, 20]. Therapeutic options for SS are extremely limited. In recent years, good results have been reported from the application of the immunobiological agent Anakinra - an inhibitor of IL-1 [4, 10, 12, 18, 20-23].
A case with a delayed diagnosis of Schnitzler syndrome, partially influenced by systemic corticosteroids is herein reported.
Clinical case
We present a 63-year-old patient with a 5-year history of a recurrent, moderately itchy urticarial rash, distributed on the trunk and limbs, sometimes accompanied by fever up to 38.8º C, pain in the knee, ankle and elbow joints, for which he had been repeatedly hospitalized in dermatological, internal, rheumatic and allergological departments. Systemic therapy with non-steroidal anti-inflammatory agents, corticosteroids and antihistamines with a temporary response had been administered. His medical history was complicated with accompanying scoliosis, lumbar disc herniation, right kidney cyst, chronic gastritis, and benign prostatic hyperplasia.
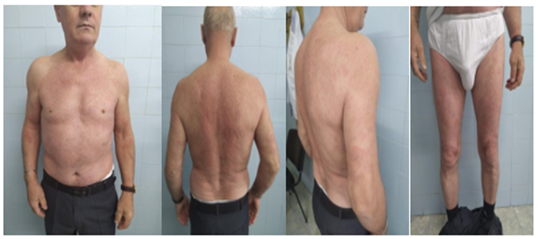
Figure 1: Disseminated urticarial lesions on the trunk and extremities
Physical examination demonstrated disseminated urticarial lesions on the skin of the trunk and extremities (Figure 1). Leukocytosis and elevated CRP proved general inflammatory constellation CRP 62 mg/dl, leukocytosis 14.6 X 10⁹/l). High levels of alpha 1, alpha 2- and beta 2- micro globulins, and a wide band of Ig M kappa class paraprotein was detected on immune electrophoresis. Numerous studies were conducted, including thyroid hormones, hepatitis B, hepatitis C, Lyme disease, leishmaniosis, Marseilles fever, brucellosis, intestinal helminths, protozoa, toxoplasmosis, echinococcosis, trichinosis, fasciola hepatica, chlamydia trachomatis, which were negative. Normal values of anti-n RNP/Sm, Sm,SS–A, SS-B, Scl-70, Jo-1, PCNA; ds DNA, rheumatoid factor, lupus anticoagulant, and cryoglobulins ruled out connective tissue disease. Thorough neoplastic work-up was negative, including tumor markers and intact positron emission tomography findings. There were no deviations in computer tomography images of the chest, abdomen and small pelvis.
Based on the adapted criteria of the SS experts from Strasbourg, we diagnosed Schnitzler syndrome. Methylprednisolone therapy was started with gradual dose reduction. At the routine one-month follow up the patient had a partial response.
Discussion
Schnitlzer's syndrome is a rare disease, with as yet incompletely elucidated pathogenesis and pathophysiology. One hypothesis suggests that IgM paraprotein synthesis stimulates the immune complexes formation and activates the complement system [7, 10, 24]. Other mechanism suggests uncontrollable activation of IL-1α [13, 25]. Some authors classify the disease as an autoinflammatory syndrome similar to cryopyrin-related periodic syndrome [3-5, 9, 12, 13, 18, 20, 25], which has been induced by NF-κβ activation [26]. SS is also considered a "paraneoplastic phenomenon" because 15-20% of patients develop lympho-proliferative disorder [4, 12, 13, 15-18]. Genetic predisposition links the disease to NLRP3 mutation [11, 13, 26], which triggers the myeloid cell population to produce abnormally high amounts of IL-1β. The latter induces chronic stimulation and influence of B cells for IgM expression [27].
Diagnosis of SS is difficult, often delayed [28], as in our patient.
Table 1: SS diagnostic criteria (Strasbourg experts' meeting)
| Main criteria | Chronic urticarial exanthema | Monoclonal IgM or IgG gammopathy | ||
| Secondary criteria | Recurrent fever > 38ºC | Impaired bone remodeling processeswith/without pain | Neutrophilic dermal infiltrate in skin biopsy | Leukocytosis > 10000/mm³ and/or CRP > 30mg/L |
For the purpose of timely and correct diagnosis, D. Lipsker and co-authors [3] introduced diagnostic criteria, which are subsequently modified by H. D. De Koning and co-authors [4] and revised at the SS experts' meeting in Strasbourg (12) (Table 1). Diagnosis is certain for 2 major and at least 2 minor criteria for IgM or 3 minor criteria for IgG and likely for 2 major and at least 1 minor criteria for IgM or 2 minor criteria for IgG [8].
Our patient meets two major and two minor diagnostic criteria: urticarial rash, monoclonal IgM gammopathy, recurrent fever, leukocytosis and increased CRP.
SS is suspected in patients with chronic urticarial rash associated with fever, fatigue, general malaise, pain in the joints, muscles or bones, lymphadenopathy, hepato-/splenomegaly, leukocytosis, increased levels of inflammatory markers. The presence of only one of the above factors may corresponds to a monoclonal dyscrasia. The manifestations of the individual symptoms vary: chronic urticaria – 100%, itching – 45-79%, fever – 88-93%, arthralgia / arthritis – 77-94%, bone pain – 68-79%, weight reduction – 64%, lymphadenopathy – 44-85%, hepatosplenomegaly – 29-34%, angioneurotic edema – 5%, leukocytosis – 69-85%, anemia – 56%, increased ESR – 95-98%, monoclonal gammopathy – 100%, IgM – 86-97% [29].
The urticarial rash is usually the first and constant manifestation of the SS. The trunk, limbs, palms and soles, head and neck are affected. It lasts about 12-24 hours, and may disappear or persist. Fever is present in the majority of patients and is ≥ 38.5 °C (88-93% of all patients). The frequency of relapses varies, and may occur daily or several times per year [2, 12-14, 29, 30].
Monoclonal gammopathy (usually IgM, less often IgG) is mandatory, but does not always occur early in the disease, sometimes delays with a few decades [31].
Before making a diagnosis of SS, it is necessary to exclude the following diseases: Still's disease in adults; cryopyrine-associated syndromes (Muckle-Wells syndrome); urticarial vasculitis; cryoglobulinemic vasculitis; systemic lupus erythematosus; hypocomplement-associated vasculitis; chronic idiopathic urticaria; lymphoproliferative diseases; monoclonal gammopathy and monoclonal gammopathy of unknown origin.
The SS is chronic relapsing condition with a variable prognosis. About a quarter of patients develop lympho-proliferative disorders such as Waldenström's macroglobulinaemia, lymphomas, leukemias, multiple myeloma, etc. [4, 12, 13, 15-18]. In a small series of 20 patients, Socumbi et al [5] reported almost half of the patients evolving into hematological dyscrasia. AA-amyloidosis occurred in 3 cases as a result of the persistent systemic inflammation [4, 19, 20].
SS patients should be strictly monitored by controlling the inflammatory activity and severity of monoclonal gammopathy [2, 12-14]. Markers of inflammation (ESR, leukocytes, CRP) need to be tested every 3 months in active disease, and twice per year - in remission. IgM titers are checked every 3 months. An increase in their levels and / or an onset of peripheral lymphadenopathy serves as an indicator for hematological disorder and necessitates immediate bone marrow puncture and lymph node biopsy.
Therapeutic options for SS are limited. No improvement was found with administration of antihistamines and thalidomide [3, 4]. Arthralgia is well alleviated with short courses of NSAIDs, especially ibuprofen [12]. Methotrexate, azathioprine, cyclosporine, intravenous immunoglobulins and plasmapheresis are ineffective [3, 4, 12]. Therapy with colchicine, dapsone, pefloxacin, hydroxychloroquine, ciclosporin, interferon-α and corticosteroids have partial effect [2, 4,7, 12, 13].
In patients with significant impairment in quality of life and/or persistence of inflammation, even in the absence of significant symptoms, administration of an IL-1 inhibitor, Anakinra 100 mg/morning, subcutaneously [12, 21-23] is recommended. Neel et al. [20] presented a retrospective analysis of 29 patients receiving Anakinra who experienced complete remission in 83% and partial remission in 17%. No lympho-proliferative malignancy was detected at 9.5 years mean observation time. In the absence of an effect of Anakinra, the diagnosis should be confirmed once again, and subsequently adjust the treatment with an increase in the dose to 200-300 mg and complimentary use of colchicine, as an adjuvant. Alternatively, Tocilizumab may be used [31]. In remission for more than 2 years, stopping treatment or continuing with colchicine for 3 to 6 months is prevented. Long-term inhibitors of IL-1 – Rilonacept [32) and Canakinumab [33-37] have been administered to patients with SS, but there is no long-term experience of their administration. There are single, conflicting reports of Rituximab and phototherapy [18]. IL-6 inhibitor was tried in non-compliant with the IL-1 inhibitor [38].
Conclusion
Schnitzler syndrome is a rare auto inflammatory disease that represents an enormous diagnostic challenge. The nonspecific clinical findings delay its verification for years. The pathogenesis is still obscured with genetic predisposition and defective innate immunity considered the most important triggers. The role of monoclonal gammopathy is not yet fully understood, most likely underlying the development of lymphoproliferative malignancies. IL-1 receptor blockers prove to be the most effective therapeutic modality. Timely diagnosis and appropriate treatment require multidisciplinary approach and deductive insight to further elucidate the scientific research in this widely expanding area of clinical medicine.
References
- Schnitzler L. (1972). Lésions urticariennes chroniques permanentes (erythème pétaloide?) Cas Clinique. J Dermatol Angers, 46:44-46.
Publisher | Google Scholor - Gusdorf L, Lipsker D. (2017). Schnitzler syndrome: a review. Curr Rheumatol Rep., 19:46-51.
Publisher | Google Scholor - Lipsker D, Veran Y, Grunenberger F et al. (2001). The Schnitzler Syndrome - four new cases and review of the literature. Medicine (Baltimore), 80:37-44.
Publisher | Google Scholor - De Koning HD, Bodar EJ, van der Meer JW. (2007). Schnitzler Syndrome Study Group. Schnitzler Syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment. Semin Arthritis Rheum., 37:137-148.
Publisher | Google Scholor - Sokumbi O, Drage LA, Peters MS. (2012). Clinical and histopathologic review of Schnitzler syndrome: the Mayo Clinic experience (1972-2011). J Am Acad Dermatol.; 67: 1289-1295.
Publisher | Google Scholor - Lipsker D, Spehner D, Drillien R et al. (2000). Schnitzler syndrome: heterogeneous immunopathological findings involving IgM-skin interactions. British J Dermatol., 142:954-959.
Publisher | Google Scholor - Asli B, Bievenu B, Cardoliani F et al. (2007). Chronic Urticaria and Monoclonal IgM gammopathy (Schnitzler syndrome). Report of 11 cases treated with Pefloxacin. Arch Dermatol., 143:1046-1050.
Publisher | Google Scholor - Eiling E, Schroder JO, Gross WL et al. (2008). The Schnitzler syndrome: chronic urticaria and monoclonal gammopathy — an autoinflammatory syndrome? J Dtsch Dermatol Ges.; 6: 626-631.
Publisher | Google Scholor - Tinazzi E, Puccetti A, Patuzzo G et al. (2011). Schnitzler syndrome, an autoimmune-autoinflammatory syndrome: report of two new cases and review of the literature. Autoimmun Rev.; 10:404-409.
Publisher | Google Scholor - Zuberbier T, Maurer M. Urticarial Vasculitis and Schnitzler Syndrome. Immunol Allergy Clin N Am. 2014; 34:141-147.
Publisher | Google Scholor - Rowczenio DM, Pathak S, Arostegui JI et al. (2018). Molecular genetic investigation, clinical features, and response to treatment in 21 patients with Schnitzler syndrome. Blood, 131:874-881.
Publisher | Google Scholor - Simon A, Asli B, Braun-Falco M et al. Schnitzler Syndrome: diagnosis, treatment, and follow-up. Allergy. 2013; 68:562-568.
Publisher | Google Scholor - Lipsker D. (2010). The Schnitzler syndrome. Orphanet J Rare Dis., 38-45.
Publisher | Google Scholor - De Koning HD. (2014). Schnitzler’s syndrome: lessons from 281 cases. Clin Transl Allergy, 4:41-56.
Publisher | Google Scholor - Schnitzler L, Schubert B, Boasson M et al. (1974). Urticaire chronique, lesions osseuses, macroglobulinemie IgM maladie de Waldenstrom. Bull Soc Franc Derm Syph., 81:363-367.
Publisher | Google Scholor - Lim W, Shumak KH, Reis M et al. (2002). Malignant evolution of Schnitzler’s syndrome: chronic urticaria and IgM monoclonal gammopathy: report of a new case and review of the literature. Leukemia & Lymphoma, 43:181-186.
Publisher | Google Scholor - Dalle S, Balme B, Sebban C et al. (2006). Schnitzler syndrome associated with systemic marginal zone B-cell lymphoma. Br J Dermatol., 155:827-829.
Publisher | Google Scholor - Eiling E, Moller M, Kreiselmaier I et al. (2007). Schnitzler syndrome: treatment failure to rituximab but response to anakinra. J Am Acad Dermatol., 57:361-364.
Publisher | Google Scholor - Claes K, Bammens B, Delforge M et al. (2008). Another devastating complication of the Schnitzler syndrome: AA amyloidosis. Br J Dermatol., 158:182-184.
Publisher | Google Scholor - Néel A, Benoit H, Barbarot S et al. (2014). Long term effectiveness and safety of interleukin-1 receptor antagonist (anakinra) in Schnitzler’s syndrome. A French multicentric study. Autoimmun Rev., 13:1035-1041.
Publisher | Google Scholor - Martinez-Taboada VM, Fontalba A, Blanco R et al. (2005). Successful treatment of refractory Schnitzler syndrome with anakinra: comment on the article by Hawkins et al. Arthritis Rheum., 52:2226-2227.
Publisher | Google Scholor - Gran JT, Midtvedt Q, Haug S et al. (2011), Treatment of Schnitzler syndrome with anakinra: report of three cases and review of the literature. Scand J Rheumatol.; 40:74-79.
Publisher | Google Scholor - Billey T, Beldjerd M, Popa L et al. (2010). Schnitzler syndrome: a dramatic improvement with anakinra. Presse Med., 39:1338-1339.
Publisher | Google Scholor - De Castro FR, Masouye I, Winkelmann RK et al. (1996). Urticarial pathology in Schnitzler’s (hyper-IgM) syndrome. Dermatology., 193:94-99.
Publisher | Google Scholor - Saurat JH, Schifferly J, Steiger G et al. (1991). Anti-interleukin-1 alpha autoantibodies in humans: characterization, isotype distribution and receptor-binding inhibition-higher frequency in Schnitzler’s syndrome (urticaria and macroglobulinemia). J Allergy Clin Immunol., 88:244-256.
Publisher | Google Scholor - Bauernfeind FG, Horvath G, Stutz A et al. (2009). Cutting edge: NF-κβ activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol., 183:787-797.
Publisher | Google Scholor - De Koning HD, van Gijn ME, Stoffels M et al. (2015). Myeloid lineage-restricted somatic mosaicism of NLRP3 mutations in patients with variant Schnitzler syndrome. J Allergy Clin Immunol., 135:561-564.
Publisher | Google Scholor - Jain T, Offord CP, Kyle RA et al. (2013). Schnitzler syndrome: an under-diagnosed clinical entity. Haematol., 98(10):1581-1585.
Publisher | Google Scholor - Vanderschueren S, van der Veen A. (2017). The Schnitzler syndrome: chronic urticaria in disguise: a single-centre report of 11 cases and a critical reappraisal of the literature. Clin Exp Rheumatol., 35:69-73.
Publisher | Google Scholor - Gusdorf L, Asli B, Barbarot S et al. (2017). Schnitzler syndrome: validation and applicability of diagnostic criteria in real-life patients. Allergy, 72:177-182.
Publisher | Google Scholor - Varella TC, Nishimura MY, Machado MC et al. (2005). Schnitzler’s syndrome without monoclonal gammopathy. Acta Derm Venereol., 85:272-273.
Publisher | Google Scholor - Krause K, Weller K, Stefaniak R et al. (2012). Efficacy and safety of the interleukin-1 antagonist rilonacept in Schnitzler syndrome:an open-label study. Allergy, 67:943-950.
Publisher | Google Scholor - De Koning HD, Schalkwijk J, van der Meer JW, et al. (2011). Successful canakinumab treatment identifies IL-1β as a pivotal mediator in Schnitzler syndrome. J Allergy Clin Immunol, 128:1352-1354.
Publisher | Google Scholor - Vanderschueren S, Knockaert D. (2013). Canakinumab in Schnitzler syndrome. Semin Arthritis Rheum, 42:413-416.
Publisher | Google Scholor - De Koning HD, Schalkwij J, Van der Ven-Jongerkriig J et al. (2013). Sustained efficacy of the monoclonal anti-interleukin-1 beta antibody canakinumab in a 9-month trial in Schnitzler’s syndrome. Ann Rheum Dis., 72:1634-1638.
Publisher | Google Scholor - Pesek R, Fox R. (2014). Successful treatment of Schnitzler syndrome with canakinumab. Cutis, 94:E11-E12.
Publisher | Google Scholor - Krause K, Tsianakas A, Wagner N, et al. (2016). Efficacy and safety of canakinubab in Schnitzler syndrome: a multicenter randomized placebo-controlled study. J Allergy Clin Immunol, S0091-6749;16:30968-X.
Publisher | Google Scholor - Krause K, Feist E, Fiene M et al. (2012). Complete remission in 3 of 3 anti-IL-6-treated patients with Schnitzler syndrome. J Allergy Clin Immunol, 129:848-850.
Publisher | Google Scholor












