

Research article
Relationship Between Long Arm Duplication of X Chromosome and Lysosomal Storage Disease
- Osman Demirhan 1
Department of Medical Biology, Faculty of Medicine, Çukurova University, Balcali-Adana, Turkey
*Corresponding Author: Osman Demirhan, Department of Medical Biology, Faculty of Medicine, Çukurova University, Balcali-Adana, Turkey.
Citation: Demirhan O. (2023). Synthesis, Relationship Between Long Arm Duplication of X Chromosome and Lysosomal Storage Disease, Clinical Interventions and Clinical Trials, BRS Publishers. 1(1); DOI: 10.59657/2993-1096.brs.23.003
Copyright: © 2023 Osman Demirhan, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: February 28, 2023 | Accepted: May 19, 2023 | Published: May 25, 2023
Abstract
Lysosomal storage disorders (LSDs) are rare genetic and clinically heterogeneous metabolic disorders. Despite years of work on the genetic and molecular basis of these diseases, little is still known about their pathology. However, it is mostly inherited autosomal recessively, but three show X-linked inheritance. Copies of the long arm of the X chromosome are very rare, especially in males. Here, we reported a duplication of the q22.3 region of the X chromosome in a male individual in cytogenetic analysis with peripheral blood lymphocyte cultures. Our Finding shows that although the history of Xq duplications in men is limited, as confirmed in the literature review, the duplication in the current case may be associated with LSD and may be helpful to better understand the expression of X-linked traits.
Keywords: long arm; x chromosome; lysosomal storage disease; inherited autosomal
Introduction
Lysosomal storage disorders are a rare, genetically and clinically heterogeneous group of metabolic disorders. Mutations in lysosomal genes cause gradual accumulation of substrate, cell dysfunction, and cell death within the lysosome. Lysosomal storage diseases affect approximately 1 in 5,000-8,000 individuals worldwide. Most of these diseases are autosomal recessive inherited more than 50 disease groups. Currently, it is known that mutations in more than 70 genes disrupt lysosomal metabolism, leading to neuropathological effects, musculoskeletal abnormalities, dysmorphia, and seizure formation. Diagnosis of LSDs is made according to tests such as clinical symptoms, enzymatic analysis and single gene sequencing [1]. The most common lysosomal storage disorder is Gaucher disease. Most LSDs are inherited in an autosomal recessive fashion. Only three of these diseases; The disease Fabry disease, Hunter disease, and Danon disease are inherited as an X-linked recessive [2]. Fabry disease is caused by mutations in the GLA gene located in the q22.1 region of the X chromosome, resulting in a mutant α-galactosidase-A enzyme. In the literature, cytogenetic findings of this disease are almost nonexistent. The available studies are only molecular findings. In this study, we present a male individual with lysosomal storage disease with de novo Xq duplication, defined cytogenetically as 46, XY, dup (X) (q22.3), and review the literature.
Materials and Methods
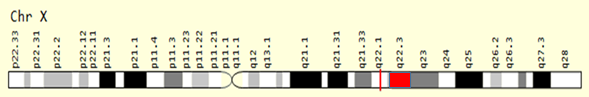
Metaphase chromosome analysis from peripheral blood lymphocyte cultures and GTG-banding were performed using standard protocols. The karyotype was interpreted as 46, XY, dup (X) (q22.3) (Figure 1). The maternal karyotype was normal, indicating a de novo origin for the dup(X) in the proband.
Chromosome
Discussion
The study of chromosomes or cytogenetic analysises are a reliable way to help diagnose diseases, plan treatment, or evaluate the effectiveness of treatment. We report a male patient with lysosomal storage disease with duplication of the long arm of the X chromosome at region 22.3. This observed chromosomal aberration is the first cytogenetic finding found in lysosomal storage patients. It is important in this respect. LSDs are generally inherited autosomal recessively, but only three are X-linked recessively (q22.1 region). In the present patient, the duplication on the long arm of the X chromosome appears to be consistent with the X-linked recessive inheritance pattern of the disease. Chromosome Xq duplication is the presence of an extra copy of the genetic material found on the long arm of the X chromosome in each cell.Intrachromosomal duplication or unbalanced X/Y or X/autosome translocation can cause Xq duplications. Duplicates can affect phenotype by changing gene dosage, i.e. extra genes lead to excess proteins. We could not find a similar case in the literature. At the same time, the disease-associated X-linked recessive gene is located in the q22.1 region, whereas we found the damaged region in the q22.3 region of the chromosome. It is quite interesting that the damaged region is in an advanced region of the known chromosome region. In this case, it is thought that there may be new genes related to the disease in the X(q22.3) region or that the duplication found may affect the expression of the known related gene in the q22.1 region.In addition, duplication increases the expression of the gene/genes while reducing how it causes disease or that possible gene/genes in this region may be permutated.
Xq duplications refer to chromosomal disorders resulting from involvement of the long arm of the X chromosome. Prevalence of Xq duplications remains unknown. In general, long arm copies of the X chromosome are rare in males. In most cases, the chromosomal abnormality is X-linked and often inherited from the mother. Classical X-linked disorders are usually a vertical form of inheritance, in which heterozygous females pass the allele on to their sons and daughters. Daughters of affected men are always heterozygous, and sons of affected men are always normal. The vast majority of mutations in X-linked genes cause disease in males only. However, many of the X-linked diseases show different rates of penetrance and expressivity in both genders. In some conditions, such as Fabry disease, heterozygotes are usually affected. The a-galactosidase A gene consists of seven exons located on the X (q22.1). Over 250 mutations have been described in all seven exons, the majority of which are missense point mutations. Isolated duplications of the long arm of the X chromosome are rare, and approximately 60 cases have been reported in the literature [3,4]. The abnormal phenotypes associated with these duplications are usually associated with gene dosage in the duplication site. Most of these were duplications of the Xq12→ q24 or Xq26.3→ qter region [5-13]. Among these duplications, Xq21→ Xq24 duplications were reported in two patients and proximal duplication in three male patients: Xq21→ q22 [14-18].
Bulduğumuz duplikasyon da bu bölgeler arasında yer alıyor. Clinical manifestations widely vary depending on the gender of the patient and on the gene content of the duplicated segment. Dysfunction of genes can affect the phenotype. In general, males are typically more severely affected than females. Chromosome Xq duplication syndromes are typically inherited in an X-linked fashion and affect new-born children. This syndrome is a rare congenital defect and symptoms may appear at birth or afterward. Men with duplications of the long arm of the X chromosome often have significant mental retardation and birth defects resulting from functional disomy of the duplicated regions. Most females with Xq copies have normal phenotypes. However, this damage is detected after the birth of an abnormal boy. Such children have been reported in females with phenotypic abnormalities such as short stature, microcephaly, developmental delay/mental retardation, and gonadal dysgenesis. The causes of variability in phenotype have been reported to be due to the size or location of the copied segment, random X inactivation, duplication of dose-sensitive genes and genes that normally escape inactivation, and incomplete inactivation of a portion of the replicated segment [19,20].Although each LSD results from mutations in a different gene and deficiency in enzyme activity or protein function, all LSDs share a common feature in that they result in the accumulation of normally degraded substrates within lysosomes. Many of the LSD genes have been characterized and it is assumed that there are different mutations, in part, within the same gene. However, genotype-phenotype correlations are not always valid. For example, in Gaucher disease, sometimes there may be significant differences in the clinical picture of the disease between siblings, and in some cases one sibling is severely affected while the other has almost no signs of the disease [21]. Apart from genetic and environmental factors, it is thought that some other factors probably play a role in the progression of the disease. While most X-linked diseases show different rates of penetration and expression in both sexes, the vast majority of mutations in X-linked genes cause disease in males only [22].
Conclusion
Although date on Xq duplications in males are limited, there appears to be an association between duplication and lysosomal storage disease, as demonstrated in the present case. However, it can be said that there may be a relationship between genes in the Xq2.23 region and LDS. Our results may help to elucidate the lysosomal mechanisms in the pathogenesis of the disease and to better understand the expression of X-linked traits.
References
- Parenti G, Andria G, Ballabio A. (2015). Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med. 66:471-486.
Publisher | Google Scholor - Matte U, Baldo G, Giugliani R. (2011). Non Viral Gene Transfer Approaches for Lysosomal Storage Disorders. In: Non-Viral Gene Therapy. InTech.
Publisher | Google Scholor - Lammer EJ, Punglia DR, Fuchs AE, Rowe AG, Cotter PD. (2001). Inherited duplication of Xq27.2qter: Phenocopy of infantile Prader–Willi syndrome. Clin Dysmorphol. 10:141-144.
Publisher | Google Scholor - Armstrong L, McGowan-Jordan J, Brierley K, Allanson JE. (2003). De novo dup(X)(q22.3-q26) in a girl with evidence that functional disomy of X material is the cause of her abnormal phenotype. Am J Med Genet. 116:71-76.
Publisher | Google Scholor - Thode A, Partington MW, Yip MY, Chapman C, Richardson VF, Turner G. A (1988). new syndrome with mental retardation, short stature and an Xq duplication. Am J Med Genet. 30:239-250.
Publisher | Google Scholor - Carrio A, Soler A, Perez MT, Margarit E, Salami C. et.al. (1991). Prenatal diagnosis of dup(X)(q13-q22) in CVS. Clin Genet. 40:12.
Publisher | Google Scholor - Muscatelli F, Verna JM, Philip N, Moncla A, Mattei MG. et.al. (1992). Physical mapping of an Xq-proximal interstitial duplication in a male. Hum Genet. 88:691-694.
Publisher | Google Scholor - Yokoyama Y, Narahara K, Tsuji K, Moriwake T, Kanzaki S. Et.al. (1992). Growth hormone deficiency and empty sella syndrome in a boy with dup(X)(q13.3-q21.2). Am J Med Genet. 42:660-664.
Publisher | Google Scholor - Miller PR, Williams LJ, McKeown C. (1993). A case of Xq duplication in a growth retarded male infant. Clin Cytogenet Bull. 2:46.
Publisher | Google Scholor - Vasquez AI, Rivera H, Bobadilla L, Crolla JA. (1995). A familial Xp chromosome, dup(Xq26.3-qter). J Med Genet. 32:891-893.
Publisher | Google Scholor - Apacik C, Cohen M, Jakobeit M, Schmucker B, Schuffenhauer S. (1996). Thurn und Taxis E, Genzel-Boroviczeny O, Stengel-Rutkowski S. Two brothers with multiple congenital anomalies and mental retardation due to disomy (X)(q12;q13.3) inherited from the mother. Clin Genet. 50:63-73.
Publisher | Google Scholor - Goodman BK, Shaffer LG, Rutberg J, Leppert M, Harum K. et.al. (1998). Inherited duplication Xq27-qter at Xp22.3 in severely affected males: Molecular cytogenetic evaluation and clinical description in three unrelated families. Am J Med Genet. 80:377-384.
Publisher | Google Scholor - Novelli A, Bernardini L, Salpietro DC, Briuglia S, Merlino MV. et.al. (2004). Disomy of distal Xq in males: Case report and overview. Am J Med Genet. 128:165-169.
Publisher | Google Scholor - Steinbach P, Horstmann W, Scholz W. (1980). Tandem duplication dup(X)(q13q22) in a male proband inherited from the mother showing mosaicism of X-inactivation. Hum Genet. 54:309-313.
Publisher | Google Scholor - Cremers FP, Pfeiffer RA, van de Pol TJ, Hofker MH, Kruse TA. et.al. (1987). An interstitial duplication of the X chromosome in a male allows physical fine mapping of probes from the Xq13-q22 region. Hum Genet. 77:23-27.
Publisher | Google Scholor - SchmidtM, Du Sart D, Kalitsis P, LevershaM, Dale S. et.al. (1991). Duplications of the X chromosome in males: Evidence that most parts of the X chromosome can be active in two copies. Hum Genet. 86:519-521.
Publisher | Google Scholor - Sparkes RS, Salter WJ, Blaker RG, Muller HM. (1997). Insertional trans location into the X chromosome of a 46,XY male. Clin Genet. 12:114-118.
Publisher | Google Scholor - Schwartz S, Schwartz MF, Panny SR, Peterson CJ, Waters E. et.al. (1986). Inherited X-chromosome inverted tandem duplication in a male traced to a grandparental mitotic error. Am J Hum Genet. 38:741-750.
Publisher | Google Scholor - Aughton DJ, AlSaadi AA, Johnson JA, Transue DJ, Trock GL. (1993). Dir dup(X)(q13→ qter) in a girl with growth retardation, microcephaly, developmental delay, seizures, and minor anomalies. Am J Med Genet. 46:159-164.
Publisher | Google Scholor - Zhang A, Weaver DD, Palmer CG. (1997). Molecular cytogenetic identification of four X chromosome duplications. Am J Med Genet. 68:29.
Publisher | Google Scholor - Beutler E, Grabowski GA. Gaucher disease. (1995). In: Scriver CR, Beaudet AC, Sly WS, Valle D, eds. The Metabolic and Molecular Basis of Inherited Disease. Vol 2. 7th ed. New York, NY: McGraw-Hill Book Co. 2641-2670.
Publisher | Google Scholor - Willard HF. (2000). The sex chromosomes and X chromosome inactivation. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Vogelstein B (eds) The metabolic and molecular bases of inherited Disease, 8th ed. McGraw-Hill. 1191-1221.
Publisher | Google Scholor












