

Review Article
Nonclassic Congenital Adrenal Hyperplasia and Female Infertility: Pathogenesis, Diagnosis and Contemporary Management
1Shanxi Medicine University, Taiyuan, Shanxi, China.
2Shanxi Provincial Children's Hospital (Shanxi Provincial Maternal and Child Health Hospital), Taiyuan, Shanxi, China.
3Center of Reproductive Medicine, Children’s Hospital of Shanxi and Women Health Center of Shanxi, Taiyuan, Shanxi, China.
*Corresponding Author: Xueqing Wu, Shanxi Provincial Children's Hospital (Shanxi Provincial Maternal and Child Health Hospital), Taiyuan, Shanxi, China and Center of Reproductive Medicine, Children’s Hospital of Shanxi and Women Health Center of Shanxi, Taiyuan, Shanxi, China.
Citation: Wang S, Li X, Zhang N, Zhang X, Wu X. (2026). Nonclassic Congenital Adrenal Hyperplasia and Female Infertility: Pathogenesis, Diagnosis and Contemporary Management, Journal of Women Health Care and Gynecology, BioRes Scientia Publishers. 6(1):1-15. DOI: 10.59657/2993-0871.brs.26.105
Copyright: © 2026 Xueqing Wu, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: January 12, 2026 | Accepted: February 02, 2026 | Published: March 02, 2026
Abstract
Congenital adrenocortical hyperplasia (CAH) is an autosomal recessive disorder caused by deficiencies in enzymes involved in adrenal steroid hormone biosynthesis. This enzymatic defect impairs cortisol and aldosterone production, accompanied by hyperandrogenemia, disrupted progesterone (P) metabolism, and other clinical and biochemical abnormalities. CAH comprises classic and nonclassic (NCCAH) subtypes. Women with NCCAH often present with hyperandrogenic manifestations (e.g., hirsutism, acne, menstrual disorders) that may lead to anovulatory infertility, while some have subtle or no clinical symptoms, resulting in underdiagnosis. This review systematically discusses the pathogenesis, clinical manifestations, diagnosis, differential diagnosis, reproductive endocrine impacts, genetic testing, and treatment strategies for infertility in women with NCCAH, aiming to provide individualized clinical guidance for managing these infertile patients.
Keywords: congenital adrenocortical hyperplasia; nonclassic congenital adrenal hyperplasia; infertility; 21-hydroxylase deficiency; reproductive endocrine disorder
Introduction
CAH is one of the most common autosomal recessive disorders, with a global prevalence of 1 in 10,000 to 1 in 16,000-exceeding that of cystic fibrosis and phenylketonuria-and is classified as a rare disease. NCCAH, the milder subtype, has a higher prevalence (up to 1 in 500) with substantial ethnic and regional variations [1]. The clinical manifestations and age of onset of these two subtypes in female patients vary considerably. Women of reproductive age with NCCAH often present with reproductive dysfunction, a condition that is easily overlooked in clinical practice. Pregnancy rates in women with NCCAH seeking treatment for infertility or hyperandrogenemia range from 65% to 95 percentage across studies, reflecting variations in sample size, diagnostic criteria, and study design [2-4] (Table 1). A Swedish cohort study involving 272 women with CAH due to 21-hydroxylase deficiency (21-OHD) and 27,200 sex-, age-, and birthplace-matched controls demonstrated that the fertility rate in women with CAH (25.4%) was significantly lower than that in the control group (45.8%) (P less than 0.001) [5]. However, research on infertility in women with NCCAH remains limited globally. The actual fertility rate is difficult to determine, as mild cases are often undiagnosed. This review summarizes the current evidence on infertility in women with NCCAH, focusing on pathogenesis, diagnosis, and treatment. We propose an integrated management strategy involving specialized clinical care, patient education, and genetic counseling to optimize pregnancy outcomes, addressing the gap in comprehensive clinical guidance for this underdiagnosed population.
Table 1: Comparison of Pregnancy Rates, Miscarriage Rates, and Management Strategies in Women with NCCAH.
| Author (Year) | Sample Size | Pregnancy Rate (Before/After Treatment) | Miscarriage Rate (Before/After Treatment) | Key Management Strategies |
| Moran et al. (2006) | 101 women, 203 pregnancies | Before Dx: 68.8% (term delivery) After Dx: 86.2% (term delivery) | Before Dx: 25.4% After Dx: 6.2% (P less than 0.002) | Preconception screening and diagnosis; glucocorticoid therapy to reduce miscarriage risk |
| Bidet et al. (2010) | 190 women, 187 pregnancies† | Overall:90.5% of women achieved pregnancy Subgroup (GC-treated): 77 pregnancies | Without GC: 26.3% With GC: 6.5% (P less than 0.01) | Hydrocortisone therapy to normalize androgen levels and menstrual cycles; recommend treatment for women seeking pregnancy |
| Eyal et al. (2017) | 75 women, 187 pregnancies‡ | Overall: 96 percentage achieved pregnancy No significant difference in TTC with/without GC | Overall: 20.3% No significant difference between treated and untreated | Glucocorticoids may shorten time to conceive in subfertile women; dose tapering during pregnancy based on hormone levels |
Table Footnotes: †187 total pregnancies (190 women minus 3 not specified). ‡187 total pregnancies (75 women with multiple pregnancies included). Abbreviations: AI: Adrenal Insufficiency; CAH: Congenital Adrenal Hyperplasia; Dx: Diagnosis; GC: Glucocorticoid; HC: Hydrocortisone; TTC: Time to Co.
Clinical Phenotypes of CAH
The onset and severity of clinical manifestations are determined by the degree of cortisol synthesis reduction, androgen hypersecretion, age of onset, and severity of CYP21A2 mutation [1]. CAH is classified into three subtypes with distinct fertility outcomes: salt-wasting CAH (SW-CAH) has a fertility rate of 0-10%, simple virilizing CAH (SV-CAH) has a sixfold higher rate (33-55%), and NCCAH is associated with a fertility rate of 63-90% [6].
Salt-Wasting CAH (SW-CAH): This is the most severe form of classic CAH, typically caused by a severe deficiency in 21-hydroxylase (21-OH). It accounts for 65-75% of CAH cases. It is characterized by adrenal insufficiency secondary to marked deficiencies in cortisol and aldosterone, presenting with hypovolemic shock, hyponatremia, hyperkalemia, metabolic acidosis, and sometimes hypoglycemia [7], along with virilization of the external genitalia in females. If left untreated, this condition can be life-threatening during the neonatal period. The residual enzyme activity in SW-CAH is typically less than 1%.
Simple Virilizing CAH (SV-CAH): This is a milder form of classic CAH, accounting for 25-35% of classic CAH cases. It is usually detected in the prepubertal period through clinical signs such as hyperandrogenism, precocious puberty, premature pubarche, clitoromegaly, rapid growth, accelerated skeletal maturation, adrenal hyperplasia, and body odor. Most women exhibit subclinical cortisol deficiency [8,9]. Women with this type have sufficient residual 21-OH activity to avoid salt wasting, but inadequate cortisol synthesis leads to excessive androgen levels. The residual enzyme activity for this form of CAH is 1-2%.
NCCAH or Late-Onset CAH: NCCAH is the mildest and most common subtype of CAH, caused by mild-to-moderate 21-hydroxylase deficiency (21-OHD) with an adolescent onset. It is characterized by hyperandrogenic manifestations, including hirsutism (60-78%), acne (33%), menstrual cycle disturbances (55%), and reduced fertility (12%) [10,11]. These clinical features are more pronounced in females; males are often asymptomatic or present only with acne and/or reduced fertility [12]. The overall prevalence of NCCAH among women presenting with hyperandrogenism is approximately 4% [13]. Residual enzyme activity ranges from 20% to 60%.
Pathogenesis of NCCAH
NCCAH is characterised by impaired synthesis of adrenal corticosteroids. The most common cause is 21-OHD due to variants in the CYP21A2 gene8, accounting for approximately 95% of all NCCAH cases [14]. Other causes of NCCAH include deficiencies in 11β-hydroxylase (11β-OH), 3β-hydroxysteroid dehydrogenase (3β-HSD), 17α-hydroxylase/17-20 lyase, cytochrome P450 oxidoreductase (POR), steroidogenic acute regulatory (StAR) protein, and P450 cholesterol side-chain cleavage enzyme (P450scc) (Table 2) [9]. The clinical manifestations vary depending on the type and severity of the enzyme deficiency.
Table 2: Enzyme Deficiencies Associated with NCCAH.
| Phenotype | Hormone Profile | Biochem Block | Enzyme | Gene |
| Postnatal hyperandrogenism, hirsutism, acne, oligomenorrhea, infertility | 17-OHP↑, androstenedione↑, testosterone↑ | Impaired conversion of progesterone to DOC and 17-OHP to 11-deoxycortisol | 21-hydroxylase (21-OH) | CYP21A2 |
| Hypertension, hypokalemia, mild virilization, menstrual irregularities | 11-hydroxyandrostenedione↑, DOC↑, androgens↑; renin↓ | Impaired conversion of 11-deoxycortisol to cortisol and DOC to corticosterone | 11β-OH | CYP11B1 |
| Rare nonclassic form with hirsutism, oligomenorrhea | 17-OHP↑, DHEA↑ | Impaired conversion of pregnenolone to progesterone and DHEA to androstenedione | 3β-HSD Type II | HSD3B2 |
| Rare nonclassic form with oligomenorrhea, ovarian cysts, infertility | Progesterone↑, corticosterone↑, DOC↑; cortisol and sex steroids↓ | Impaired conversion of pregnenolone to 17-hydroxypregnenolone and DHEA to androstenedione | 17α-hydroxylase/17-20 lyase | CYP17A1 |
| Ambiguous genitalia, labial fusion, clitoromegaly, skeletal anomalies | Progesterone↑, 17-OHP↑; cortisol and sex steroids↓ | Impaired electron transfer to microsomal P450 enzymes | P450 oxidoreductase | POR |
| May lead to adrenal insufficiency | Deficiency of all adrenal and gonadal steroids | Impaired cholesterol transPORt into mitochondria | StAR protein | StAR |
| May lead to adrenal insufficiency | Deficiency of all adrenal and gonadal steroids | Impaired conversion of cholesterol to pregnenolone | P450scc | CYP11A1 |
Table Footnotes: Abbreviations: 17-OHP = 17-Hydroxyprogesterone; 3β-HSD = 3β-hydroxysteroid dehydrogenase; DHEA = Dehydroepiandrosterone; DOC = Deoxycorticosterone; P450scc = P450 cholesterol side-chain cleavage enzyme; StAR = Steroidogenic Acute Regulatory Protein.; ↑ = increased hormone level; ↓ = decreased hormone level.
21-OHD
21-OHD has a prevalence of 1 in 200 in the U.S. population [15]. In over 90% of 21-OHD cases, residual enzyme activity ranges from 20% to 50% [16]. This enzymatic defect blocks the conversion of progesterone (P) to deoxycorticosterone (DOC) and 17-hydroxyprogesterone (17-OHP) to 11-deoxycortisol, leading to reduced cortisol and aldosterone synthesis (Figure 1). This disruption impairs negative feedback regulation, increasing adrenocorticotropic hormone (ACTH) and corticotropin-releasing hormone (CRH) secretion and promoting the accumulation of steroid precursors. Consequently, excessive androgen production induces hyperandrogenemia, while elevated P levels further contribute to metabolic dysfunction [17]. Clinically, these abnormalities manifest as disorders of sexual development, menstrual irregularities, anovulation, and persistently elevated P levels-all of which can lead to infertility and exert significant long-term impacts on physical and psychological health [18].
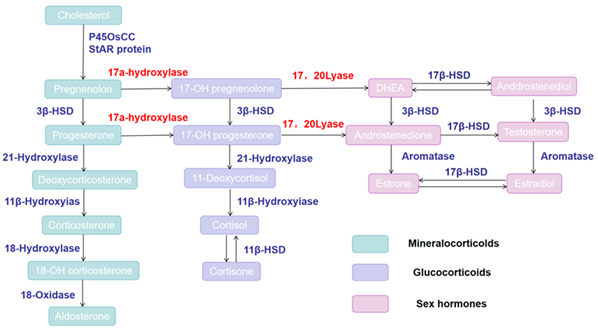
Figure 1: Schematic of the adrenal seroidogenesis pathway.
11β-OH Deficiency(11β-OHD)
11β-OH catalyzes the final step of cortisol synthesis in the adrenal zona fasciculata, converting 11-deoxycortisol to cortisol and 11-deoxycorticosterone to corticosterone [19]. Variants in the CYP11B1 gene lead to impaired enzyme activity, resulting in reduced cortisol production and accumulation of precursor steroids, including 11-deoxycortisol and 11-deoxycorticosterone (Figure 1) [20]. Deficiency of this enzyme may lead to sodium retention, hypokalemia, and hypertension, alongside clinical manifestations such as virilization in females and ambiguous genitalia in female infants. This condition is highly similar to NCCAH caused by 21-OHD deficiency and is thus easily misdiagnosed. Hypertension represents a key distinguishing feature between 11β-OHD and 21-OHD deficiency.
3β-HSD Deficiency
Type II 3β-hydroxysteroid dehydrogenase (3β-HSD) is encoded by the HSD3B21 gene on chromosome 8q2120.3β-HSD deficiency-related CAH is caused by nonsense variants, missense variants, small deletions, or splice-site variants, and is primarily rePORted in the Jewish population [20]. Once produced, pregnenolone and dehydroepiandrosterone (DHEA) are converted to P and androstenedione, respectively, by 3β-HSD, a critical step in steroid synthesis (Figure 1). Deficiency in 3β-HSD leads to the accumulation of precursors such as pregnenolone and DHEA, reducing the synthesis of hormones including P and androstenedione. This can cause abnormal sexual development, ovulatory dysfunction, and impaired fertility.
17α-hydroxylase/17-20 lyase Deficiency (17-OHD)
17-OHD is an autosomal recessive disorder caused by mutations in the CYP17A1 gene located at 10q24.3. The gene product, cytochrome P450c17, is expressed in both adrenal and gonadal tissues and exhibits two enzymatic activities: 17α-hydroxylase and 17,20-lyase [14,21]. Accordingly, mutations in CYP17A1 can result in three phenotypic variants: combined 17α-hydroxylase/17,20-lyase deficiency (the most common), isolated 17α-hydroxylase deficiency, or isolated 17, 20-lyase deficiency [14]. Biochemically, this enzymatic defect impairs the synthesis of cortisol and sex hormones, leading to the accumulation of mineralocorticoid precursors-particularly deoxycorticosterone (DOC) (Figure 1). Excess DOC induces hypertension and hypokalemia. The estimated incidence of 17-OHD ranges from 1 in 50,000 to 1 in 100,000 [22-24]. Typical clinical features include primary amenorrhea and absent secondary sexual characteristics. Some patients may present with spontaneous but oligomenorrheic menstruation, ovarian cysts, and infertility [24,25]. Concurrent glucocorticoid deficiency is also observed. For affected individuals, achieving a live birth typically requires assisted reproductive technologies (ART).
POR Deficiency (PORD)
The POR gene encodes cytochrome P450 oxidoreductase, a key enzyme that transfers electrons from NADPH to cytochrome P450 (CYP450) enzymes to support their catalytic functions. Variants in POR impair the activity of multiple CYP450 enzymes involved in steroidogenesis, including 17α-hydroxylase and 21-OH, leading to disrupted biosynthesis of cortisol, aldosterone, and sex steroids [26]. The biochemical hallmark of PORD includes reduced production of cortisol and sex steroids, with shunting of precursors into alternative metabolic pathways. Clinically, PORD presents with a broad phenotypic spectrum. Prenatally, it is associated with skeletal anomalies such as craniosynostosis, radiohumeral synostosis, and midface hypoplasia, resembling Antley-Bixler syndrome (ABS)9. Postnatally, during infancy or childhood, the most common manifestations of PORD include ambiguous genitalia-sometimes associated with Prader grade V virilization, labial fusion, clitoromegaly, vaginal atresia, and a single urogenital orifice [27].
StAR Deficiency and P450scc Deficiency
StAR protein transPORts cholesterol into mitochondria, while P450scc converts cholesterol to pregnenolone-both representing essential initial steps in the steroidogenic pathway (Figure 1). StAR deficiency causes severe adrenal insufficiency accompanied by lipid accumulation in adrenal glands. Since StAR is not expressed in the placenta, affected 46, XX individuals survive birth but subsequently develop sex hormone deficiency [28]. In contrast, P450scc is expressed in all steroidogenic tissues, including the placenta. Deficiency of this enzyme is usually lethal in utero due to placental P insufficiency. Most reported survivors are 46, XY individuals with complete sex reversal and adrenal insufficiency [29].
Impact of NCCAH on Reproductive Fertility
Fertility in women with NCCAH is lower than in the general population, with 10%-30% of reproductive-aged patients reporting infertility [3,10,30]. Multiple studies confirm reduced pregnancy or live birth rates in reproductive-aged women with 21-OHD [31]. Over 30%-50% of women with NCCAH exhibit substantial ovulatory and menstrual dysfunction [10,32]. An international multicenter study of 101 women with NCCAH across 203 pregnancies reported pregnancy complication rates of 2.5% for classic CAH and 14.8% for NCCAH5.The causes of infertility in these women are multifactorial, including but not limited to congenital malformations of the reproductive tract, impaired follicular development, and decreased endometrial receptivity due to abnormal P and T secretion26.Additionally, comorbidity with polycystic ovary syndrome (PCOS), alterations in sexual behavior, and psychosocial factors also contribute [33-37].
Hormonal factors play a pivotal role in conception and pregnancy maintenance. Increased synthesis and secretion of adrenal androgens and progesterone significantly impact fertility in women with CAH [38]. Elevated androgens and/or progesterone induce endometrial molecular changes that disrupt embryo implantation. Inadequate glucocorticoid (GC) regulation in NCCAH leads to adrenal androgen excess, which suppresses pituitary gonadotropin secretion, inhibiting follicular development and ovulation, thereby causing infertility [39]. Androgen excess also exerts a direct effect on the ovary, inhibiting late follicular development and further compromising ovulation [39,40]. Another contributor to reduced fertility in women with NCCAH is elevated progesterone concentrations, which alter endometrial receptivity and fallopian tube motility, leading to ovulatory disorders [41]. Excessive adrenal-derived progesterone production reduces LH pulse frequency. Low LH/FSH ratios indicate inadequate control of adrenal androgens. In women, the menstrual cycle (including ovulation) is dependent on LH/FSH balance; thus, insufficient LH/FSH signaling results in amenorrhea, oligomenorrhea, abnormal ovulation induction [42], and disruption of endometrial thickening, embryo implantation, and sperm migration. Conversely, chronic exposure to excess adrenal androgens can impair hypothalamic sensitivity to progesterone, subsequently leading to excessive LH secretion. Therefore, in a subset of women, neuroendocrine dysfunction in 21-OHD may involve increased gonadotropin-releasing hormone (GnRH) pulse frequency, resulting in LH levels exceeding FSH levels. This contributes to an ovarian cyst phenotype32, accompanied by irregular menstrual cycles.
Despite androgen and progesterone excess, most women with NCCAH can conceive spontaneously, and pregnancy rates among women with NCCAH and infertility are comparable to those in the general population [2,43,44]. Bidet et al. rePORted that 57% of pregnancies occurred spontaneously without any treatment, and 83% of women who conceived did so within one year. Although pregnancy rates in women with NCCAH are similar to those in the general population [43,44], these women experience higher miscarriage rates than healthy women. This may be attributed to luteal phase dysfunction in women with NCCAH [45]. A US study comparing pregnancy outcomes between the general population and women with CAH (as part of a larger adrenal insufficiency cohort, including 32 with classic CAH) reported higher rates of miscarriage (43.8%), cesarean delivery (64.3%), and preterm birth (32.3%) [5,46,47]. Furthermore, emerging evidence suggests that the risk of infertility increases with age. The rate of singleton live births is significantly higher in women with NCCAH who receive pre-pregnancy counseling from experienced reproductive endocrinologists and undergo targeted treatment compared to those who conceive spontaneously [48].
Diagnosis and Differential Diagnosis of NCCAH with Infertility
NCCAH can be diagnosed at any age but is most commonly identified post-pubertally, during infertility evaluations, or following recurrent miscarriages [17]. Compared to classic CAH, the clinical manifestations of NCCAH are atypical, highly heterogeneous, and often subtle. Consequently, it is not routinely diagnosed during the neonatal period or early childhood, leading to underdiagnosis and delayed intervention. The diagnostic workflow for NCCAH is outlined below:
History Taking
Women with NCCAH are prone to underdiagnosis due to atypical clinical manifestations. Most women with NCCAH present to reproductive endocrinology clinics with complaints of menstrual irregularities, hyperandrogenic features (e.g., hirsutism, acne), and infertility. For suspected cases, a detailed menstrual history should be obtained, including cycle regularity, duration of menstrual flow, and age at menarche. The presence of hyperandrogenic symptoms-such as acne, hirsutism, and alopecia-should be systematically assessed. A comprehensive family history should also be elicited, particularly regarding infertility, adrenal disorders, early-onset hirsutism, or other endocrine abnormalities.
Physical Examination
A thorough physical examination should include assessments of body hair distribution, skin condition, and body mass index (BMI). Due to excessive postnatal androgen exposure, women with NCCAH often exhibit varying degrees of hyperandrogenic manifestations, such as acne, hirsutism, male-pattern hair loss, deepened voice, and increased muscle mass. Some individuals may also present with underdeveloped secondary sexual characteristics, primary amenorrhea, or oligomenorrhea. For women seeking fertility, a pelvic examination is indicated to evaluate ovarian size and morphology, as well as signs of PCOS (e.g., menstrual irregularities, hirsutism, acne) and female virilization.
Laboratory Testing
Measurement of Serum 17-OHP Levels: According to the 2018 Clinical Practice Guideline published by The Endocrine Society, the diagnosis of NCCAH is typically based on basal and/or ACTH-stimulated levels of 17-OHP. It is recommended that blood samples for women suspected of NCCAH be collected in the morning after an overnight fast. If the patient is menstruating, testing should be performed during the early follicular phase of the ovarian cycle (days 3 to 5) to minimize the impact of cyclic variations16. The normal reference range for serum 17-OHP in adult women during the follicular phase is 200-1000 ng/dL (6-30 nmolA level > 1000 ng/dL (> 30 nmol/L) is diagnostic of NCCAH, while a level less than 200 ng/dL (less than 6 nmol/L) essentially rules out the diagnosis [48,49]. However, sole reliance on 17-OHP cut-off values may miss some NCCAH cases [50], so clinical guidelines recommend confirmatory testing with liquid chromatography-tandem mass spectrometry (LC-MS/MS) [49].
ACTH Stimulation Test (Syntropin Stimulation Test): The rate of underdiagnosis for NCCAH may be relatively high due to mild or nonspecific clinical manifestations and baseline 17-OHP levels that potentially fall within the normal range. For women with mildly elevated baseline 17-OHP (2.0-10.0 ng/mL [6-30 nmol/L]), the diagnosis of NCCAH can be confirmed by assessing the 17-OHP response to ACTH stimulation. During the test, the baseline 17-OHP level is measured first. Subsequently, 250 μg of ACTH is administered via intramuscular or intravenous injection1, followed by repeat measurements of serum 17-OHP levels at 30- and 60-minutes post-injection. A significant elevation in stimulated 17-OHP-for example, exceeding 10 ng/mL (30 nmol/L)-confirms the diagnosis [16], though specific diagnostic criteria may vary by laboratory. The ACTH stimulation test is a robust tool for diagnosing NCCAH, but its interpretation requires careful correlation with clinical manifestations and other laboratory findings.
Androgen Level Testing: Measurements of various androgens must be interpreted against age-, sex, and pubertal stage-specific normal reference ranges. Serum levels of free T and total T should be measured. Women with NCCAH may exhibit mild to moderate elevations in androgens. Assessment of other androgen metabolites-such as androstenedione and dehydroepiandrosterone sulfate (DHEAS)-may also be indicated. However, DHEAS can also be elevated in PCOS, limiting its diagnostic specificity for NCCAH.
Other Hormone Level Testing: Levels of LH, FSH, cortisol, and estrogens should be measured to evaluate the patient’s ovulatory function and hormonal feedback mechanisms. Plasma renin concentration (PRC) or renin activity (PRA), angiotensin II, and aldosterone may be elevated; however, these markers lack high diagnostic specificity for NCCAH.
Imaging Studies: Timely imaging studies (adrenal ultrasound, computed tomography [CT]) are recommended to differentiate adrenal tumors or other adrenal pathologies. Pelvic ultrasound of the uterus and adnexa is also indicated to evaluate ovarian morphology and rule out concurrent PCOS.
Differential Diagnosis: The differentiation between NCCAH and PCOS represents a common clinical challenge due to their overlapping features of hyperandrogenism, ovulatory dysfunction, and metabolic complications. It is estimated that approximately 33% of women diagnosed with PCOS actually have NCCAH [51]. Both conditions may present with elevated basal androstenedione and T levels [52], but PCOS more frequently exhibits an elevated LH/FSH ratio [53,54]. In contrast, NCCAH is characterized by significantly elevated basal or ACTH-stimulated 17-OHP levels, with post-ACTH 17-OHP exceeding 10 ng/mL (30 nmol/L) being diagnostic. Given that misdiagnosis can lead to inappropriate treatment-such as managing PCOS with oral contraceptives or metformin while neglecting the glucocorticoid therapy required for NCCAH-targeted screening with the ACTH stimulation test is essential in high-risk scenarios [48]. These high-risk groups include women with early-onset hyperandrogenism, a family history of CAH, specific ethnic backgrounds, or poor response to conventional PCOS therapies. Accurate biochemical confirmation, supplemented by genetic testing when indicated, is critical to ensure specific and effective intervention-particularly for optimizing fertility outcomes.
Molecular Genetic Testing and Genotyping for NCCAH Variants
Genetic testing serves as a critical diagnostic tool for NCCAH. A strong correlation exists between genotype severity and the degree of ovarian dysfunction and infertility [16]. It can be utilized to identify CYP21A2 genotypes and heterozygous carriers when biochemical test results are inconclusive or when preconception genetic counseling is indicated48. Additionally, genetic testing is essential for confirming diagnoses in challenging cases, acts as a key determinant of prognosis, and facilitates accurate genetic counseling [55].
Prenatal Genetic Diagnosis
For couples with a family history of NCCAH, prenatal genetic counseling can help them understand the disease’s inheritance pattern and associated risks, thereby facilitating informed family planning decisions. CAH exhibits a strong genotype-phenotype correlation; by contrast, this correlation is weaker in NCCAH. Therefore, the clinical management of NCCAH should be primarily based on clinical manifestations and hormonal test data [56]. With technological advances over recent decades, more precise prenatal diagnostic methods have been developed, which can be categorized as invasive and non-invasive types.
Invasive Testing Methods: Amniocentesis entails the acquisition of fetal DNA from amniotic fluid under ultrasound guidance, using a 22-gauge spinal needle via the abdominal approach, and is typically performed at 12-14 weeks of gestation. Fetal hormone analysis of amniotic fluid was the primary method for prenatal diagnosis. Fetal cells obtained via this procedure were initially used for HLA typing (given that CYP21A2 is located within the HLA complex) to determine the inheritance of maternal and paternal haplotypes, and can also be applied for genetic analysis. Chorionic villus sampling (CVS) involves the collection of placental villi under ultrasound guidance, with a needle or specialized catheter inserted via the cervical or abdominal route to obtain fetal DNA; this procedure is typically performed at 10-11 weeks of gestation. Currently, these two invasive techniques are available in numerous countries and clinical centers [57]. Although amniocentesis and CVS enable the definitive diagnosis of CAH, they are associated with potential risks including infection, bleeding, and even miscarriage [58].
Non-Invasive Testing Methods: The advent of non-invasive prenatal testing (NIPT) in 2011 enabled non-invasive screening for CAH, a technique that isolates fetal cell-free DNA (cfDNA) from maternal peripheral blood [54]. Unlike intact fetal cells, cfDNA is rapidly cleared from the maternal circulation shortly after delivery [59,60], thereby eliminating diagnostic interference in subsequent pregnancies. NIPT may evolve into the new standard diagnostic modality in the future [61]; however, its application for NCCAH detection remains limited to date [62,63]. Furthermore, cfDNA extraction and analysis facilitate the diagnosis of multiple genetic disorders, including X-linked disorders, CAH, and fetal sex determination. This technology is particularly advantageous for the evaluation of male fetuses, as fetal sex can be identified as early as the 5th week of gestation via polymerase chain reaction (PCR) targeting Y-chromosome-specific sequences [60,64]. Following fetal sex determination and CYP21A2 genotyping, prenatal intervention can be considered to prevent external genital virilization in affected female fetuses. In contrast, prenatal treatment is not indicated for 46, XY fetuses, as virilization-related complications do not arise in this population [65]. Currently, non-invasive prenatal screening methods tailored specifically to NCCAH are not yet fully mature, but cfDNA-based non-invasive genetic testing platforms hold promise for future development.
Preimplantation Genetic Diagnosis
Preimplantation genetic testing (PGT) is available in many countries for families at risk of severe genetic disorders, including 21-OHD. Combined with in vitro fertilization (IVF), PGT enables genetic screening of embryos for CYP21A2 gene variants before uterine implantation. During IVF, when embryos develop to the blastocyst stage, several cells can be biopsied for genetic analysis without compromising normal embryonic development. The preferred sampling method for PGT is trophectoderm biopsy of day 5-6 blastocysts, which typically contain approximately 120 cells. This approach enables isolation of the inner cell mass (ICM)-the cell population that gives rise to the fetus-from the trophectoderm, with 5-10 cells required for reliable genotyping. Embryos confirmed to be free of CYP21A2 variants can then be selected for implantation, thereby reducing the genetic risk of NCCAH in offspring. While this strategy enables ultra-early genetic diagnosis, it carries a potential risk of damaging the developing embryo [17].
NCCAH is an autosomal recessive disorder; thus, affected individuals may be either homozygous (harboring identical variants on both alleles) or compound heterozygous (carrying two distinct variants, one on each allele). Approximately 70% of women with NCCAH are compound heterozygotes, with different pathogenic variants on each allele [49]. Notably, around two-thirds of these women carry one allele associated with CAH4. The NCCAH phenotype largely correlates with the residual enzymatic activity encoded by the milder variant. It has been reported that 25-50% of women with NCCAH carry mild variants on both alleles [32,66]. Theoretically, women with NCCAH have a 1/350 risk of having a child with classic CAH without prior CYP21A2 genotyping. A retrospective French study of 190 women with NCCAH estimated this risk to be 1.5% [4]. The estimated carrier frequency of NCCAH ranges from 1:7 to 1:16. The epidemiology of NCCAH remains less well-defined than that of classic CAH, as NCCAH is not detected by routine newborn screening programs [15]. Therefore, preconception CYP21A2 genotyping is recommended to accurately stratify individual reproductive risks. Given the potential for pregnancy complications and adverse effects on offspring, preconception genetic testing and counseling with a specialist in high-risk obstetrics are strongly recommended for women with a confirmed diagnosis of CAH [67].
Treatment for NCCAH with Infertility
Standardized treatment has significantly improved fertility outcomes in women with NCCAH complicated by infertility. In one study, the singleton live birth rate was higher among women diagnosed and treated for NCCAH prior to conception than among those who conceived spontaneously (86% vs. 69%, respectively), with no significant differences in the rates of ectopic pregnancy, preterm birth, stillbirth, or twin/multiple gestations [3]. It has been reported that 25% of pregnancies ended in spontaneous miscarriage prior to NCCAH diagnosis, whereas this rate decreased to 10% in women who received a confirmed diagnosis and initiated treatment before pregnancy [3] a finding indicating that timely diagnosis and treatment enhance pregnancy success rates in this patient population.
The primary treatment goals for these women include individualized glucocorticoid administration to control excessive adrenal androgens while minimizing long-term adverse effects, fertility management combined with genetic counseling, and optimization of patient quality of life (Figure 2). Given that excessive adrenal progesterone secretion during the follicular phase is a key contributor to infertility in NCCAH, progesterone levels should be maintained below 0.6 ng/mL (2 nmol/L) throughout the treatment course1. Although fertility is impaired in most women with NCCAH, successful pregnancy is achievable with appropriate clinical management, which is critical to prevent irreversible adverse outcomes for the patient’s pregnancy, delivery, and embryonic/fetal development.
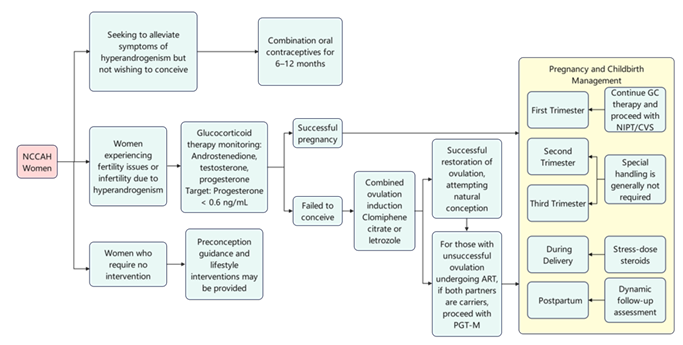
Figure 2: Management algorithm for infertility in women with NCCAH.
Oral Contraceptives (OCs) and Anti-Androgen Therapy
For women with NCCAH exhibiting signs of androgen excess (e.g., hirsutism, menstrual irregularities) who do not desire fertility, the typical first-line treatment is combination oral contraceptives for 6-12 months. The therapeutic mechanisms of oral contraceptives include the following: a) Suppression of LH and androgen production to reduce hyperandrogenemia; b) Stimulation of hepatic sex hormone-binding globulin (SHBG) synthesis, thereby lowering free androgen concentrations; c) Mild reduction in adrenal androgen synthesis; d) Decreased binding of androgens to their receptors; e) Increased clearance of T; f) Mild inhibition of 5α-reductase in the pilosebaceous unit, the enzyme that catalyzes the conversion of T to the most potent androgen, dihydrotestosterone (DHT)48. For women with a poor response to treatment, anti-androgens such as spironolactone (initial dose 50-100 mg/day) or low-dose flutamide (62.5-125 mg/day) can be added to the regimen1. Due to their suppression of ACTH and ovarian/adrenal androgens, OCs are a first-line therapy for improving hyperandrogenism and oligomenorrhea.
Glucocorticoid Replacement Therapy
Although glucocorticoid therapy can suppress pituitary ACTH secretion and reduce adrenal androgen production, most women with NCCAH do not have clinically significant glucocorticoid deficiency. Therefore, glucocorticoid treatment is generally not recommended, as it exerts suppressive effects on the HPA axis [3]. Furthermore, lifelong exposure to supraphysiological glucocorticoid doses can lead to severe cardiovascular, metabolic, and skeletal complications, and may also exert adverse effects on patients’ mental health and quality of life [56,68]. During acute illness, exogenous glucocorticoid administration can induce iatrogenic adrenal insufficiency, which carries the risk of life-threatening Addisonian crisis—a severe complication that is rarely observed in untreated women with NCCAH [69].
However, updated clinical guidelines identify a specific subgroup for whom glucocorticoid therapy is indicated: adult females with atypical nonclassic NCCAH presenting with patient-concerning hyperandrogenism or infertility [70]. If such women desire fertility, they may first attempt natural conception, and glucocorticoid treatment is also unnecessary following conception. Given that 10%-30% of women with NCCAH report subfertility [3], glucocorticoid therapy can be considered for this cohort to help reduce exposure to elevated adrenal androgens and restore normal ovulatory cycles [71-73], thereby shortening the time to pregnancy. Glucocorticoids suppress excessive secretion of CRH and ACTH from the hypothalamus and pituitary gland, restore physiological LH/FSH secretion, and inhibit adrenal overproduction of androgens and progesterone to improve fertility outcomes [15]. Administration of hydrocortisone, prednisone, or prednisolone before and during pregnancy can enhance reproductive outcomes, with conception rates approaching 90% [2,45]. Both hydrocortisone and prednisone/prednisolone are inactivated by placental 11β-HSD2, which protects the fetus from potentially supraphysiological maternal glucocorticoid exposure. In contrast, dexamethasone should be avoided, as it readily crosses the fetoplacental barrier [73]. Notably, glucocorticoid administration throughout pregnancy has been reported to reduce miscarriage rates [9].The recommended treatment regimens for adult women with NCCAH-related hyperandrogenism who desire fertility are as follows: hydrocortisone (15-25 mg/day, divided into 2-3 daily doses), prednisolone (4-6 mg/day, divided into 2 daily doses), prednisone (5-7.5 mg/day, divided into 2 daily doses), or dexamethasone (0.25-0.5 mg/day, once daily), all of which demonstrate comparable efficacy [10,16]. Real-world observational studies reveal substantial variability in glucocorticoid regimens, with doses often exceeding the recommended range [72,74]. Nonetheless, glucocorticoids should be administered on an individualized and judicious basis to restore ovulatory cycles and optimize fertility in this patient population [69].
Preconception: For women at genetic risk of NCCAH, cfDNA-based non-invasive genetic testing can be performed as early as 6 weeks of gestation. This allows for timely diagnosis and intervention prior to the onset of fetal genital organogenesis (week 9 of gestation) [54,71], with the primary goal of suppressing fetal adrenal androgen production and preventing external genital virilization in female fetuses [75]. Treatment should be initiated with prednisone at a dosage of 4-5 mg/day, administered in divided doses, and can be increased to 7.5 mg/day based on the requirements for maintaining normal ovulatory function. Treatment should be discontinued if the fetus is identified as male or an unaffected female; otherwise, it should be continued at a dose tailored to the mother’s pre-pregnancy weight until term delivery.
During Pregnancy: Dosing adjustments for hydrocortisone therapy during pregnancy should not be guided by plasma renin activity levels, as this parameter is physiologically elevated during gestation. Instead, T and androstenedione concentrations should serve as the primary monitoring markers [76,77].
- First Trimester (less than12 weeks): No hydrocortisone dose adjustment is typically required during the first trimester [12]. If GC therapy was initiated prenatally, it should be continued. Non-invasive prenatal testing (NIPT) is recommended during this period, and chorionic villus sampling (CVS) is a viable option for prenatal genetic diagnosis.
- Second Trimester (12-24 weeks): For women receiving GC therapy, close monitoring for adrenal crisis is indicated. The mother should be assessed for signs of adrenal insufficiency during pregnancy (e.g., orthostatic hypotension). If an adrenal crisis develops, urgent intervention is required, with administration of stress-dose steroids and intravenous fluids following the same protocol used for non-pregnant individuals [78]. Amniocentesis can be performed at 15-20 weeks for prenatal genetic screening. Management strategies should be determined based on the results of CVS or amniocentesis; treatment should be discontinued if the fetus is confirmed to be male or an unaffected female.
- Third Trimester (24-41 weeks): During the second and third trimesters, the hydrocortisone dose may need to be increased by 25-40% to meet the elevated physiological cortisol demands of pregnancy, although a universal consensus on this dose adjustment is lacking. If an adrenal crisis occurs after fetal viability is achieved (≥ 24 weeks of gestation), maternal treatment should be accompanied by continuous fetal well-being monitoring and assessment of uterine contractions. In such cases, co-management by an experienced endocrinologist and a high-risk obstetrics specialist is recommended [75].
During Delivery: Delivery constitutes a physiological stress event equivalent to major surgery, necessitating the administration of stress-dose steroids. Hydrocortisone (100 mg intravenously [IV]) should be given at the onset of active labor, followed by either 50 mg hydrocortisone IV every 6 hours or a continuous infusion of 200 mg hydrocortisone over 24 hours throughout labor and delivery-regardless of whether the delivery is vaginal or cesarean. Concurrent intravenous fluid replacement is indicated, and continuous cardiovascular monitoring is required.
Postpartum: Stress-dose steroids should be continued immediately following delivery. Generally, the prenatal glucocorticoid dose should be doubled for 24-48 hours, then rapidly tapered to the pre-pregnancy dose and further weaned gradually over the subsequent days. Early postpartum follow-up assessments by the reproductive endocrinology team are recommended until the dose is reduced to the pre-pregnancy baseline [79]. Women with NCCAH receiving chronic glucocorticoid therapy may be at increased risk of complications such as impaired wound healing and infection. Therefore, close monitoring of the surgical incision site (if applicable) is essential to ensure adequate wound healing.
Ovulation Induction
If ovulation is not restored with glucocorticoid monotherapy, ovulation induction can be considered using agents including clomiphene citrate, aromatase inhibitors, injectable gonadotropins, and adjunctive metformin [80]. Consultation with a reproductive endocrinology and infertility (REI) specialist is strongly recommended. Patients should undergo etiological screening in accordance with standard infertility diagnosis and treatment protocols. If ovulatory dysfunction persists or other infertility etiologies are present concomitantly, ovulation-inducing medications or ART may be administered in combination to optimize the likelihood of pregnancy.
Assisted Reproductive Technology (ART)
For patients who fail to conceive with the aforementioned treatments, in vitro fertilization with embryo transfer (IVF-ET) may be pursued as a therapeutic option. Consideration should be given to a freeze-all embryo strategy, with embryo transfer scheduled in a subsequent cycle to avoid elevated progesterone concentrations induced by the IVF stimulation protocol. If both parents are CAH mutation carriers, or one parent is affected and the other is a carrier, the fetal inheritance risk is significantly increased, and PGT is essential [77,81]. The main challenges of ART in this population include androgen excess-induced anovulatory cycles and elevated follicular-phase progesterone levels, which impair endometrial receptivity, fallopian tube motility, and cervical mucus quality [41]. Concurrently, close monitoring of P levels during controlled ovarian hyperstimulation (COH) is crucial. If indicated, a freeze-all strategy should be implemented, with frozen-thawed embryo transfer (FET) timed optimally to prevent excessive progesterone from compromising endometrial receptivity. For women with this genetic family background, PGT is strongly recommended to optimize pregnancy outcomes and reduce the risk of fetal CAH inheritance.
Social and Psychological Support
Both NCCAH and infertility can impose substantial psychological stress on affected women, who may experience emotional distress including anxiety, depression, reduced self-worth, and social isolation. Recognizing these psychological impacts and providing timely, effective support is crucial; professional psychological counseling should be offered when indicated.
For women with NCCAH, we recommend collaborative management by a multidisciplinary team comprising endocrinologists, obstetrician-gynecologists, and geneticists with specialized expertise in this disorder. Preconception counseling is strongly advised for this patient cohort. Women diagnosed with infertility should receive individualized treatment tailored to the underlying etiology. Concurrently, it is essential to emphasize the value of establishing long-term follow-up registries to clarify the safety profile of prenatal treatments, develop standardized evidence-based therapeutic protocols (e.g., regarding dosage, tapering schedules, and initiation timing), and ultimately determine the feasibility of integrating prenatal therapy into routine clinical practice.
Summary and Future Perspectives
This review summarizes the impact of NCCAH on female reproductive health, with a particular focus on its association with infertility. In recent years, the prevalence of infertility among women with NCCAH has gradually increased. For NCCAH-affected women desiring fertility, initial attempts at natural conception are recommended. If spontaneous pregnancy fails, preconception genetic testing and consultation with a maternal-fetal medicine specialist are advised. Individualized and judicious glucocorticoid therapy is recommended to restore ovulatory regularity and improve fertility potential, followed by the administration of ovulation-inducing agents or gonadotropins as clinically indicated. For couples seeking to prevent the transmission of NCCAH to their offspring, management by reproductive endocrinology and infertility (REI) specialists is recommended, utilizing IVF-ET combined with preimplantation genetic testing for monogenic disorders (PGT-M).
Although the molecular mechanisms of NCCAH have been further clarified, establishing robust genotype-phenotype correlations remains challenging, as they are influenced by modifier genes, epigenetic regulators, and environmental factors. Current evidence is mostly derived from retrospective studies with small sample sizes and inconsistent diagnostic criteria, limiting the generalizability of conclusions. A deeper understanding of the endocrine physiology of the adrenal cortex, reproductive axis, and maternal-fetal interface during pregnancy is also required. Comprehensive, patient-centered care tailored to individual life stages and clinical circumstances is essential for further optimizing pregnancy outcomes in women with NCCAH. Given the fertility aspirations of many women with NCCAH, well-designed prospective randomized controlled trials are needed to evaluate the efficacy and safety of combined glucocorticoid and ovulation-inducing therapies. Additionally, long-term follow-up studies of offspring born to NCCAH mothers are required to assess the long-term impacts of prenatal interventions. Therefore, the provision of genetic counseling, prenatal diagnosis, timely clinical interventions, and evidence-based guidance throughout the infertility management process is paramount for achieving favorable maternal and fetal outcomes.
Declarations
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author Contributions
Shimin Wang prepared, drafted and revised the manuscript. Xiuping Zhang assisted with data collection and manuscript revision. Jiayao Chen and Ningxin Zhang assisted with data collection. Xueqing Wu performed manuscript review and administrative support. All authors approved the submitted version and agreed to the All authors approved the submitted version and agreed to take personal responsibility for their contributions and to ensure that any questions regarding the accuracy or completeness of the work are addressed. Any questions relating to the accuracy or completeness of the work are properly. Any issues related to the accuracy or completeness of the work are properly investigated and documented in the literature.
Funding
This work was supported by the Four batches Science and Technology Innovation Plan Project of Shanxi Reproductive and Heritage Research Science and Technology Cultivation Team Program (2020TD19); Shanxi Provincial Basic Research Programme Free Exploration Top-level Project (202203021211062).
References
- Uslar T, Olmos R, Martínez-Aguayo A, Baudrand R. (2023). Clinical Update on Congenital Adrenal Hyperplasia: Recommendations from a Multidisciplinary Adrenal Program. J Clin Med. 12(9):3128.
Publisher | Google Scholor - Eyal O, Ayalon-Dangur I, Segev-Becker A, Schachter-Davidov A, Israel S, et al. (2017). Pregnancy in women with nonclassic congenital adrenal hyperplasia: Time to conceive and outcome. Clin Endocrinol (Oxf). 87(5):552-556.
Publisher | Google Scholor - Moran C, Azziz R, Weintrob N, et al. (2006). Reproductive outcome of women with 21-hydroxylase-deficient nonclassic adrenal hyperplasia. J Clin Endocrinol Metab. 91(9):3451-3456.
Publisher | Google Scholor - Bidet M, Bellanné-Chantelot C, Galand-PORtier MB, et al. (2010). Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 95(3):1182-1190.
Publisher | Google Scholor - Hirschberg AL, Gidlöf S, Falhammar H, et al. (2021). Reproductive and Perinatal Outcomes in Women with Congenital Adrenal Hyperplasia: A Population-based Cohort Study. J Clin Endocrinol Metab. 106(2):e957-e965.
Publisher | Google Scholor - Wang F, Yu B, Yang W, Liu J, Lu J, et al. (2012). Polycystic ovary syndrome resembling histopathological alterations in ovaries from prenatal androgenized female rats. J Ovarian Res. 5(1):15.
Publisher | Google Scholor - Yoon JH, Hwang S, Kim JH, Kim GH, Yoo HW, et al. (2024). Prenatal diagnosis of congenital adrenal hyperplasia due to 21-hydroxylase deficiency through molecular genetic analysis of the CYP21A2 gene. Ann Pediatr Endocrinol Metab. 29(1):54-59.
Publisher | Google Scholor - Merke DP, Auchus RJ. (2020). Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. Ingelfinger JR, ed. N Engl J Med. 383(13):1248-1261.
Publisher | Google Scholor - El-Maouche D, Arlt W, Merke DP. (2017). Congenital adrenal hyperplasia. The Lancet. 390(10108):2194-2210.
Publisher | Google Scholor - Moran C, Azziz R, Carmina E, et al. (2000). 21-Hydroxylase-deficient nonclassic adrenal hyperplasia is a progressive disorder: a multicenter study. Am J Obstet Gynecol. 183(6):1468-1474.
Publisher | Google Scholor - Dubey S, Tardy V, Chowdhury MR, et al. (2017). Prenatal diagnosis of steroid 21-hydroxylase-deficient congenital adrenal hyperplasia: Experience from a tertiary care centre in India. Indian J Med Res. 145(2):194-202.
Publisher | Google Scholor - Falhammar H, Nordenström A. (2015). Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. 50(1):32-50.
Publisher | Google Scholor - Jha S, Turcu AF. (2021). Nonclassic Congenital Adrenal Hyperplasia: What Do Endocrinologists Need to Know? Endocrinol Metab Clin North Am. 50(1):151-165.
Publisher | Google Scholor - Albu AI, Iancu ME, Albu DN. (2023). Successful Treatment of Infertility in a Patient with Probable 17 Hydroxylase Deficiency and Particularities of Association with Adrenal Autoimmunity-A Case Report and Review of the Literature. Life. 13(4):921.
Publisher | Google Scholor - Hannah-Shmouni F, Morissette R, Sinaii N, et al. (2017). Revisiting the prevalence of nonclassic congenital adrenal hyperplasia in US Ashkenazi Jews and Caucasians. Genet Med. 19(11):1276-1279.
Publisher | Google Scholor - Speiser PW, Arlt W, Auchus RJ, et al. (2018). Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 103(11):4043-4088.
Publisher | Google Scholor - Simpson JL, Rechitsky S. (2019). Prenatal genetic testing and treatment for congenital adrenal hyperplasia. Fertil Steril. 111(1):21-23.
Publisher | Google Scholor - Turcu AF, Auchus RJ. (2015). Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. 44(2):275-296.
Publisher | Google Scholor - Turcu AF, Rege J, Auchus RJ, Rainey WE. (2020). 11-Oxygenated androgens in health and disease. Nat Rev Endocrinol. 16(5):284-296.
Publisher | Google Scholor - Krone N, Arlt W. (2009). Genetics of congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 23(2):181-192.
Publisher | Google Scholor - Kara L, Cicek D, Siraz UG, et al. (2024). Congenital Adrenal Hyperplasia with Combined 21-hydroxylase deficiency and 17α-hydroxylase/17,20-lyase deficiency: An undervirilized male. Eur J Med Genet. 69:104952.
Publisher | Google Scholor - Xu Y, Jiang S, Yan Z, et al. (2022). Phenotypic Heterogeneity and Fertility Potential of Patients With 17-Hydroxylase/17,20-lyase Deficiency. J Clin Endocrinol Metab. 107(6):e2610-e2618.
Publisher | Google Scholor - Sun M, Mueller JW, Gilligan LC, et al. (2021). The broad phenotypic spectrum of 17α-hydroxylase/17,20-lyase (CYP17A1) deficiency: a case series. Eur J Endocrinol. 185(5):729-741.
Publisher | Google Scholor - Wolthers OD, Rumsby G, Techatraisak K, Honour JW, Hindmarsh PC. (2002). 17-Hydroxylase/17,20 lyase deficiency diagnosed during childhood. Horm Res Paediatr. 57(3-4):133-136.
Publisher | Google Scholor - Marsh CA, Auchus RJ. (2014). Fertility in patients with genetic deficiencies of cytochrome P450c17 (CYP17A1): combined 17-hydroxylase/17,20-lyase deficiency and isolated 17,20-lyase deficiency. Fertil Steril. 101(2):317-322.
Publisher | Google Scholor - Gomes LG, Bachega TASS, Mendonca BB. (2019). Classic congenital adrenal hyperplasia and its impact on reproduction. Fertil Steril. 111(1):7-12.
Publisher | Google Scholor - Fan L, Ren X, Song Y, Su C, Fu J, et al. (2019). Novel phenotypes and genotypes in Antley-Bixler syndrome caused by cytochrome P450 oxidoreductase deficiency: based on the first cohort of Chinese children. Orphanet J Rare Dis. 14(1):299.
Publisher | Google Scholor - Guo X, Zhang Y, Yu Y, et al. (2022). Getting pregnant with congenital adrenal hyperplasia: Assisted reproduction and pregnancy complications. A systematic review and meta-analysis. Front Endocrinol. 13:982953.
Publisher | Google Scholor - Adachi M, Tachibana K, Asakura Y, Yamamoto T, Hanaki K, et al. (2004). Compound heterozygous mutations of cytochrome P450 oxidoreductase gene (POR) in two patients with Antley–Bixler syndrome. Am J Med Genet A. 128A(4):333-339.
Publisher | Google Scholor - Auchus RJ, Arlt W. (2013). Approach to the patient: the adult with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 98(7):2645-2655.
Publisher | Google Scholor - Kontoleon P, Ilias I, Papapetrou PD. (2003). Successful pregnancy in a woman with rare compound heterozygoticity for congenital adrenal hyperplasia; case report. Clin Exp Obstet Gynecol. 30(4):263-264.
Publisher | Google Scholor - Livadas S, Dracopoulou M, Dastamani A, et al. (2015). The spectrum of clinical, hormonal and molecular findings in 280 individuals with nonclassical congenital adrenal hyperplasia caused by mutations of the CYP21A2 gene. Clin Endocrinol (Oxf). 82(4):543-549.
Publisher | Google Scholor - Hamed SA, Attiah FA, Abd Elaal RF, Fawzy M. (2021). Behavioral assessment of females with congenital adrenal hyperplasia. Hormones. 20(1):131-141.
Publisher | Google Scholor - Kudela G, Gawlik A, Koszutski T. (2020). Early Feminizing Genitoplasty in Girls with Congenital Adrenal Hyperplasia (CAH)-Analysis of Unified Surgical Management. Int J Environ Res Public Health. 17(11):3852.
Publisher | Google Scholor - Carroll L, Graff C, Wicks M, Diaz Thomas A. (2020). Living with an invisible illness: a qualitative study exploring the lived experiences of female children with congenital adrenal hyperplasia. Qual Life Res. 29(3):673-681.
Publisher | Google Scholor - Dobrowolska-Glazar B, Honkisz I, Sulislawski J, Tyrawa K, Wolnicki M, et al. (2020). Sexual function and health status in adult patients with Congenital Adrenal Hyperplasia. J Pediatr Urol. 16(4):464.e1-464.e6.
Publisher | Google Scholor - Zainuddin AA, Grover SR, Abdul Ghani NA, et al. (2020). Health-related quality of life of female patients with congenital adrenal hyperplasia in Malaysia. Health Qual Life Outcomes. 18(1):258.
Publisher | Google Scholor - Green D, Dineen R, O’Reilly MW, Sherlock M. (2023). Fertility and pregnancy in adrenal insufficiency. Endocr Connect. 13(2):e230088.
Publisher | Google Scholor - Walters KA, Handelsman DJ. (2018). Role of androgens in the ovary. Mol Cell Endocrinol. 465:36-47.
Publisher | Google Scholor - Lim JJ, Lima PDA, Salehi R, Lee DR, Tsang BK. (2017). Regulation of androgen receptor signaling by ubiquitination during folliculogenesis and its possible dysregulation in polycystic ovarian syndrome. Sci Rep. 7(1):10272.
Publisher | Google Scholor - Chatziaggelou A, Sakkas EG, Votino R, Papagianni M, Mastorakos G. (2019). Assisted Reproduction in Congenital Adrenal Hyperplasia. Front Endocrinol. 10:723.
Publisher | Google Scholor - Al-Azemi M, Kyrou D, Kolibianakis EM, Humaidan P, Van Vaerenbergh I, et al. (2012). Elevated progesterone during ovarian stimulation for IVF. Reprod Biomed Online. 24(4):381-388.
Publisher | Google Scholor - Claahsen-van Der Grinten HL, Stikkelbroeck NMML, Sweep CGJ, Hermus ARMM, Otten BJ. (2006). Fertility in patients with congenital adrenal hyperplasia. J Pediatr Endocrinol Metab. 19(5):677-685.
Publisher | Google Scholor - Stikkelbroeck NM, Hermus AR, Braat DD, Otten BJ. (2003). Fertility in women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Obstet Gynecol Surv. 58(4):275-284.
Publisher | Google Scholor - Krishnan K, Pillai S, Vaidyanathan G. (2023). Pregnancy in a woman with congenital adrenal hyperplasia with 11-beta-hydroxylase deficiency: A case rePORt. Obstet Med. 16(1):66-68.
Publisher | Google Scholor - Badeghiesh A, Ismail S, Baghlaf H, Suarthana E, Dahan MH. (2020). Pregnancy, delivery and neonatal outcomes among women with congenital adrenal hyperplasia: a study of a large US database. Reprod Biomed Online. 41(6):1093-1099.
Publisher | Google Scholor - Bothou C, Anand G, Li D, et al. (2020). Current Management and Outcome of Pregnancies in Women With Adrenal Insufficiency: Experience from a Multicenter Survey. J Clin Endocrinol Metab. 105(8):e2853-e2863.
Publisher | Google Scholor - Carmina E, Dewailly D, Escobar-Morreale HF, et al. (2017). Non-classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency revisited: an update with a special focus on adolescent and adult women. Hum Reprod Update. 23(5):580-599.
Publisher | Google Scholor - Nácul AP, Silva ACJSRE, Yela DA, et al. (2024). Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency in women: diagnosis and treatment: Number 11 - 2024. Rev Bras Ginecol E Obstetrícia. 46.
Publisher | Google Scholor - Maffazioli GDN, Bachega TASS, Hayashida SAY, et al. (2020). Steroid Screening Tools Differentiating Nonclassical Congenital Adrenal Hyperplasia and Polycystic Ovary Syndrome. J Clin Endocrinol Metab. 105(8):e2895-e2902.
Publisher | Google Scholor - Reichman DE, White PC, New MI, Rosenwaks Z. (2014). Fertility in patients with congenital adrenal hyperplasia. Fertil Steril. 101(2):301-309.
Publisher | Google Scholor - Chanukvadze, D., Kristesashvili, J., Kvashilava, N. (2012). Correlation of biochemical markers and clinical signs of hyperandrogenism in women with polycystic ovary syndrome (PCOS) and women with non-classic congenital adrenal hyperplasia (NCAH). Iranian Journal of Reproductive Medicine, 10(4):307-314.
Publisher | Google Scholor - Loli P, Menotti S, di Filippo L, Giustina A. (2025). Non-classical congenital adrenal hyperplasia: current insights into clinical implications, diagnosis and treatment. Endocrine. 90(1):1-16.
Publisher | Google Scholor - Podgórski R, Aebisher DA, Stompor M, Podgórska D, Mazur A. (2018). Congenital adrenal hyperplasia: clinical symptoms and diagnostic methods. Acta Biochim Pol. 65(1):25-33.
Publisher | Google Scholor - Krone N, Rose IT, Willis DS, et al. (2013). Genotype-phenotype correlation in 153 adult patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency: analysis of the United Kingdom Congenital Adrenal Hyperplasia Adult Study Executive (CaHASE) cohort. J Clin Endocrinol Metab. 98(2):E346-E354.
Publisher | Google Scholor - Claahsen - Van Der Grinten HL, Speiser PW, Ahmed SF, et al. (2022). Congenital Adrenal Hyperplasia-Current Insights in Pathophysiology, Diagnostics, and Management. Endocr Rev. 43(1):91-159.
Publisher | Google Scholor - Beta J, Lesmes-Heredia C, Bedetti C, Akolekar R. (2018). Risk of miscarriage following amniocentesis and chorionic villus sampling: a systematic review of the literature. Minerva Obstet Gynecol. 70(2).
Publisher | Google Scholor - Kazmi D, Bailey J, Yau M, et al. (2017). New developments in prenatal diagnosis of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. 165(Pt A):121-123.
Publisher | Google Scholor - Lo YM. (2000). Fetal DNA in maternal plasma: biology and diagnostic applications. Clin Chem. 46(12):1903-1906.
Publisher | Google Scholor - Guetta E. (2006). Noninvasive detection of fetal sex: the laboratory diagnostician’s view. Prenat Diagn. 26(7):635-636.
Publisher | Google Scholor - Yau M, Khattab A, New MI. (2016). Prenatal Diagnosis of Congenital Adrenal Hyperplasia. Endocrinol Metab Clin North Am. 45(2):267-281.
Publisher | Google Scholor - Okpaise OO, Ahn H, Tonni G, Ruano R. (2024). Prenatal diagnosis and in utero treatment of congenital adrenal hyperplasia: An up-to-date comprehensive review. Prenat Diagn. 44(5):635-643.
Publisher | Google Scholor - New MI, Tong YK, Yuen T, et al. (2014). Noninvasive prenatal diagnosis of congenital adrenal hyperplasia using cell-free fetal DNA in maternal plasma. J Clin Endocrinol Metab. 2014;99(6):E1022-E1030.
Publisher | Google Scholor - Wright CF, Wei Y, Higgins JP, Sagoo GS. (2012). Non-invasive prenatal diagnostic test accuracy for fetal sex using cell-free DNA a review and meta-analysis. BMC Res Notes. 5(1):476.
Publisher | Google Scholor - Kazachkova N, Gontar J, Verlinsky O, Ilyin I. (2019). Successful early fetal sex determination using cell-free fetal DNA isolated from maternal capillary blood: A pilot study. Eur J Obstet Gynecol Reprod Biol X. 3:100038.
Publisher | Google Scholor - Speiser PW, Knochenhauer ES, Dewailly D, Fruzzetti F, Marcondes JA, et al. (2000). A multicenter study of women with nonclassical congenital adrenal hyperplasia: relationship between genotype and phenotype. Mol Genet Metab. 71(3):527-534.
Publisher | Google Scholor - Rama Chandran S, Loh LM. (2019). The Importance and implications of preconception genetic testing for accurate fetal risk estimation in 21-hydroxylase congenital adrenal hyperplasia (CAH). Gynecol Endocrinol. 35(1):28-31.
Publisher | Google Scholor - Pofi R, Ji X, Krone NP, Tomlinson JW. (2024). Long‐term health consequences of congenital adrenal hyperplasia. Clin Endocrinol (Oxf). 101(4):318-331.
Publisher | Google Scholor - Adriaansen BPH, Schröder MAM, Span PN, Sweep FCGJ, Van Herwaarden AE, et al. (2022). Challenges in treatment of patients with non-classic congenital adrenal hyperplasia. Front Endocrinol. 13:1064024.
Publisher | Google Scholor - Bancos I, Kim H, Cheng HK, et al. (2025). Glucocorticoid therapy in classic congenital adrenal hyperplasia: traditional and new treatment paradigms. Expert Rev Endocrinol Metab. 20(1):33-49.
Publisher | Google Scholor - Parsa AA, New MI. (2017). Steroid 21-hydroxylase deficiency in congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. 165(Pt A):2-11.
Publisher | Google Scholor - Harris A, Seckl J. (2011). Glucocorticoids, prenatal stress and the programming of disease. Horm Behav. 59(3):279-289.
Publisher | Google Scholor - Wallensteen L, Zimmermann M, (2016). Thomsen Sandberg M, et al. Sex-Dimorphic Effects of Prenatal Treatment with Dexamethasone. J Clin Endocrinol Metab. 101(10):3838-3846.
Publisher | Google Scholor - Gao Y, Wang O, Guan W, et al. (2021). Bone mineral density and trabecular bone score in patients with 21‐hydroxylase deficiency after glucocorticoid treatment. Clin Endocrinol (Oxf). 94(5):765-773.
Publisher | Google Scholor - Maher JY, Gomez-Lobo V, Merke DP. (2023). The management of congenital adrenal hyperplasia during preconception, pregnancy, and postpartum. Rev Endocr Metab Disord. 24(1):71-83.
Publisher | Google Scholor - White PC, Speiser PW. (2000). Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 21(3):245-291.
Publisher | Google Scholor - Han TS, Walker BR, Arlt W, Ross RJ. (2014). Treatment and health outcomes in adults with congenital adrenal hyperplasia. Nat Rev Endocrinol. 10(2):115-124.
Publisher | Google Scholor - Rushworth RL, Torpy DJ, Falhammar H. (2019). Adrenal Crisis. N Engl J Med. 381(9):852-861.
Publisher | Google Scholor - Reisch N. (2019). Pregnancy in Congenital Adrenal Hyperplasia. Endocrinol Metab Clin North Am. 48(3):619-641.
Publisher | Google Scholor - Van’T Westeinde A, Karlsson L, Nordenström A, Padilla N, Lajic S. (2020). First-Trimester Prenatal Dexamethasone Treatment Is Associated with Alterations in Brain Structure at Adult Age. J Clin Endocrinol Metab. 105(8):2575-2586.
Publisher | Google Scholor - Lekarev O, Lin-Su K, Vogiatzi MG. (2015). Infertility and Reproductive Function in Patients with Congenital Adrenal Hyperplasia. Endocrinol Metab Clin North Am. 44(4):705-722.
Publisher | Google Scholor




















