

Case Report
Hypoparathyroidism and Hypothyroidism Secondary to Hemochromatosis in Beta Thalassemia Major
1.Resident Physician Hayatabad Medical Complex Peshawar, Pakistan.
2.Resident Physician Lady Reading Hospital Peshawar, Pakistan.
3.Lady Reading Hospital, Internal Medicine, Pakistan.
4.Resident physician at Combine Military Hospital, Nowshera, KPK, Pakistan.
5.Medical Officer Khyber Medical Center, Pakistan.
*Corresponding Author: Noman Salih, Resident Physician Hayatabad Medical Complex Peshawar, Pakistan.
Citation: Salih N, Ghani N, Ullah H, Ali S, Amin U, et al. (2023). Hypoparathyroidism and Hypothyroidism Secondary to Hemochromatosis in Beta Thalassemia Major. International Clinical and Medical Case Reports, BioRes Scientia Publishers. 2(2):1-5. DOI: 10.59657/2837-5998.brs.23.026
Copyright: © 2023 Noman Salih, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: August 28, 2023 | Accepted: September 12, 2023 | Published: September 15, 2023
Abstract
Hemochromatosis, an iron metabolism disorder caused by iron overload, often runs in families. It leads to various issues like cardiomyopathy, liver cirrhosis, and hormonal imbalances. This article presents a unique case of secondary hemochromatosis resulting from repeated transfusions. A 15-year-old boy with severe beta-thalassemia major received about 600 units of packed red blood cells over nine years. This led to clinical hypoparathyroidism and subclinical hypothyroidism, affecting multiple organs. Elevated ferritin and transferrin levels confirmed iron buildup, visible on MRI scans in the liver, pancreas, skeleton, heart, and thyroid gland. Such widespread organ involvement due to transfusion-related hemochromatosis is rare and makes this case noteworthy, especially considering the scarcity of similar reports in Pakistan.
Keywords: hemochromatosis; iron metabolism disorder; cardiomyopathy; liver cirrhosis; hormonal imbalances
Introduction
Excessive iron levels in the body, along with the accumulation of iron in various organs, can lead to a condition known as hemochromatosis. This condition can arise due to genetic factors or secondary causes, resulting in damage to vital organs [1]. One particular genetic mutation impacting the absorption of iron in the intestines gives rise to hereditary hemochromatosis, an autosomal recessive disorder that is more prevalent among Caucasians. Notably, there have been instances documented across different regions globally wherein secondary hemochromatosis was triggered by repeated transfusions. This has been observed in patients undergoing dialysis for end-stage renal disease, as well as those with conditions like aplastic anemia and beta-thalassemia [2]. Interestingly, the impact of repeated transfusions, which lead to an excess of iron in the body, is deemed less detrimental to organ function than hereditary hemochromatosis. This is primarily due to the differing manner in which iron is distributed within the body. In cases of secondary hemochromatosis, iron tends to accumulate within reticuloendothelial cells, as opposed to the parenchymal cells where iron buildup occurs in hereditary hemochromatosis [3].
In the context of this study, we present the case of a patient diagnosed with beta-thalassemia major. Over a span of 9 years, this individual received approximately 600 units of packed red blood cells due to their condition. The patient's hospitalization was prompted by a range of interconnected health issues, including symptomatic hypocalcemia, secondary hemochromatosis, hypoparathyroidism, thyroid irregularities, and mild hepatic abnormalities. This particular case holds significant importance as it sheds light on a unique instance of secondary hemochromatosis, showcasing its manifestation through hypoparathyroidism and hypothyroidism, a presentation that, to the best of our knowledge, has not been widely reported before.
Case Presentation
A 15-year-old boy was admitted to our hospital for the evaluation of symptomatic hypocalcemia. He had been undergoing regular blood transfusions due to beta-thalassemia major, amounting to approximately 600 units of packed red blood cells. Additionally, he had been taking specialized iron-chelating medication (deferasirox 1,500 mg/day) for the past seven years to manage elevated blood ferritin levels. Despite regular follow-up visits, his hypocalcemia persisted, leading to his outpatient visit to the medical unit for further assessment and treatment.
The patient had no family history of thyroid disease, cervical irradiation, or related conditions. However, his two siblings also had beta thalassemia major. On examination, his temperature was 98.5 °F, pulse rate 92 beats per minute, and respiratory rate 23 breaths per minute. Blood pressure was recorded at 110/70 mmHg. Notably, he exhibited dark skin and slightly pale conjunctiva. His chest examination revealed normal heart sounds, clear breath sounds, and no abnormal findings. Abdominal examination indicated mild tension and slight distention, with the liver palpable about two inches below the right ribcage. No pitting edema was observed in his legs. Neurological assessments revealed positive Chvostek and Trousseau signs.
Laboratory results are summarized in Table 1. Decreased levels of iPTH (parathyroid hormone) and testosterone indicated the involvement of iron deposits in the parathyroid glands and testes, leading to hypoparathyroidism and hypogonadism. Despite treatment with calcium carbonate and alfacalcidol twice a day, 24-hour urine calcium levels (normal range: 100–300 mg/day) had risen to 750 mg/day. Thyroid function tests displayed elevated thyroid-stimulating hormone levels and low free thyroxine levels, confirming secondary hypothyroidism due to iron accumulation. Furthermore, reduced calcium and iPTH levels confirmed hypoparathyroidism resulting from iron deposition in the parathyroid glands.
Table 1: Laboratory Investigations
| Test | Reference value | Patient’s value |
| Wbcs (/µl) | 4000 - 11000 | 13000 |
| Hb (g/dl) | 11.5 - 17.5 | 8.7 |
| Platelets (/µl) | 150000 - 450000 | 178000 |
| Bun (mg/dl) | 18 – 45 | 44 |
| Creat (mg/dl) | 0.3 - 0.9 | 0.59 |
| Alt (U/L) | 10 – 50 | 89 |
| Ast (U/L) | 8 – 33 | 75 |
| ALP (U/L) | < 390> | 357 |
| Total bilirubin (mg/dl) | 0.1 – 1 | 1.4 |
| Na (mmol/L) | 135 – 150 | 131.9 |
| K (mmol/L) | 3.5 - 5.1 | 4.23 |
| Ca (mg/dl) | 8 – 10 | 4.1 |
| Ionised Ca (mg/dl) | 4.4 - 5.2 | 3.1 |
| Ferritin (ng/mL) | 24 – 336 | > 2000 |
| Transferrin saturation (%) | 20 -50 % | 81% |
| iPTH (pg/mL) | 15 – 65 | 1.51 |
| Phosphorus (mg/dl) | 3.2 - 6.3 | 6.03 |
| 25 hydoxyVitamin D (ng/mL) | >30 | 12 |
| Mg (mg/dl) | 1.7 -2.55 | 1.6 |
| Tsh ( mIU/L) | 0.3 - 4.2 | 19.7 |
| T3 (ng/mL) | 0.8 – 2 | 0.523 |
| T4 (mcg/dL) | 5.1 - 14.1 | 2.17 |
| Testosterone (ng/mL) | 2.5 - 8.5 | 0.025 |
| LDL cholesterol (mg/dl) | <100> | 54 |
| TAG (mg/dl) | <150> | 121 |
| RBS (mg/dl) | 100 – 125 | 115 |
| HbA1c (%) | <5> | 5.3 |
Abbreviations: WBC, white blood cells; Hb, hemoglobin; BUN, blood urea nitrogen; Creat, creatinine; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ALP, alkaline phosphatase; Na, sodium; K, potassium; Ca, calcium; iPTH, intact parathyroid hormone; Tsh, thyroid stimulating hormone; T3, tri-iodothyronine; T4, Thyroxin; LDL cholesterol, low density lipoprotein cholesterol; TAG, triacyl glycerol; RBS, random blood sugar; HbA1c, hemoglobinA1c; Mg, magnesium levels
Chest radiography indicated no cardiomegaly. Abdominal and pelvic ultrasonography revealed mild hepatomegaly. Given its heightened sensitivity and specificity for detecting deposits, magnetic resonance imaging (MRI) was preferred over computed tomography (CT). The MRI shown in Figure 1 disclosed diffuse low signals in the liver, pancreas, axial skeleton, thyroid gland, and myocardium sections, indicative of widespread iron overload (Fig. 2). To eliminate the possibility of polyglandular autoimmune (PGA) disorder, the patient underwent evaluations. No symptoms of adrenal insufficiency or oral candidiasis were observed. End-organ function tests (Na, K, BUN, acid/base status) were within normal limits, as serum endocrine autoantibody screening tests were unavailable. This ruled out PGA type 1.
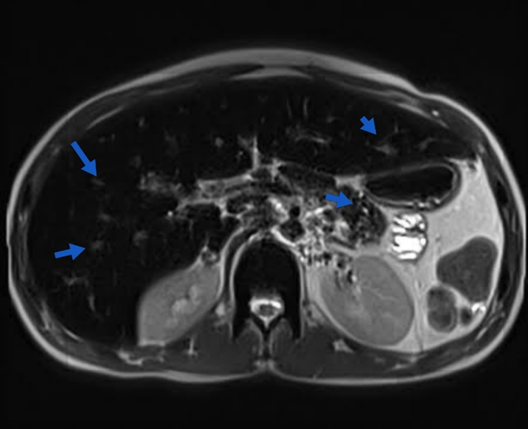
Figure 1: Blue arrows showing iron deposits in the liver and pancreas
To exclude PGA type 2, blood sugar levels and adrenal function were assessed, both yielding normal results. The absence of vitiligo or alopecia, common autoimmune disorder symptoms, was noted. Notably, the patient's blood displayed microcytic characteristics, ruling out PGA type 3, which presents with macrocytic anemia. Treatment ensued, involving increased dosages of calcium and vitamin D supplements following intravenous calcium gluconate administration. Thyroxine was prescribed for hypothyroidism, and a follow-up appointment after two weeks was scheduled. Iron depletion therapy was escalated to address hypogonadism and reduce iron stores.
Discussion
This patient's secondary hemochromatosis stems from excessive iron absorption due to frequent transfusions for beta-thalassemia major. During follow-up monitoring, he was diagnosed with hypocalcemia caused by hypoparathyroidism, subtle hypothyroidism, and liver involvement. The accumulation of iron in various organs due to heightened iron absorption can lead to organ failures, although significant instances of transfusion-induced secondary hemochromatosis are rare [3]. Hemochromatosis, whether genetic or secondary, arises from excess iron in organ tissues, damaging cells and causing organ failure [1]. Patients with hepatomegaly, skin darkening, heart disease, type 2 diabetes, arthritis, and lower urinary tract symptoms can be diagnosed if excess iron or iron infiltration with the HFE genetic mutation is confirmed via tissue biopsy [4].
Normally, the human body maintains iron levels between 3 and 4 g. Daily iron losses are around 1 mg for men and 1.5 mg for menstruating women, balanced by the same amount absorbed by the intestinal mucosa [5]. Conversely, hemochromatosis leads to the absorption of 4 mg of iron daily, raising blood iron and ferritin levels while impregnating organs. Hereditary hemochromatosis, an autosomal recessive trait associated with the HLA-A gene on chromosome 6, accounts for the majority of cases [4]. Secondary causes include excessive iron absorption due to conditions like Mediterranean anemia and sideroblastic anemia, as seen in this study [2]. Repeated transfusions, as in beta thalassemia major or aplastic anemia, can also trigger hemochromatosis. When macrophages absorb 200–250 mg of iron from a transfused unit of blood, bone marrow abnormalities prevent proper RBC production, leading to iron accumulation in organs and multiple organ failure [3].
Early hemochromatosis symptoms might include arthritis, skin discoloration, fatigue, and weakness. Excessive iron in organs like the pituitary gland, thyroid, parathyroid, liver, heart, and pancreas can result in functional impairment [6]. Liver involvement leads to hepatomegaly, elevated hepatic enzymes, cirrhosis, and hepatocellular cancer. Hemochromatosis patients have elevated mortality risks from conditions like liver cancer, cardiomyopathy, and heart failure, necessitating regular check-ups [7,8]. Diabetes results from liver and pancreatic beta-cell siderosis causing decreased insulin resistance and secretion [6]. Gonadotropin insufficiency can lead to impotence, irregular menstruation, and reduced libido [9]. While less common, siderosis can also impair the thyroid, parathyroid, and adrenal glands [10,11].
Studies on organ damage severity and duration in relation to siderosis are limited. Some studies find associations between blood transfusion quantity and serum ferritin levels, as well as endocrine issues, but more extensive studies are needed to understand these relationships better [12,13]. MRI and CT scans can assess iron impregnation without biopsy. CT is effective for diagnosing hemochromatosis when serum ferritin is over 2,000 ng/mL, and MRI is highly sensitive and specific [14,15,16,17]. In this case, MRI revealed iron overload, confirming secondary hemochromatosis [17]. Patients with transfusion-induced secondary hemochromatosis may experience severe, irreversible functional impairments [13]. Limiting transfusions, using iron-chelating meds as needed, and frequent follow-ups are crucial. Regular screenings and larger studies involving repeated transfusions are required to understand the impact on endocrine failure and other complications.
Conclusion
Individuals with secondary hemochromatosis resulting from recurrent blood transfusions in cases of beta-thalassemia major are at risk of experiencing profound functional limitations, notably disruptions in the endocrine system. Such complications can manifest as enduring issues and significantly impede daily life. Advanced imaging techniques, like MRI scans, have proven valuable in the detection of hemochromatosis-induced hypothyroidism and hypoparathyroidism. Effectively addressing and mitigating these repercussions demands a proactive approach involving regular medical evaluations, scheduled screenings, meticulous transfusion management, and the implementation of iron chelation therapy. To gain a comprehensive understanding of the interplay between transfusion frequency, duration, and the onset of endocrine insufficiencies in secondary hemochromatosis patients, further comprehensive investigations are warranted.
Acknowledgements
The authors would like to extend their sincere appreciation to the following individuals for their valuable contributions to this case report: NS: For his critical review, expert insights, and guidance throughout the research process, which greatly enriched the content and quality of this case report. NG: For his diligent efforts in data collection, meticulous analysis, and thoughtful interpretation of the clinical findings, which were instrumental in shaping the conclusions of this report. AK: For his exceptional technical expertise in medical imaging and his indispensable support in generating the visual aids and illustrations used in this case report. UA: For his assistance in conducting an extensive literature review, which provided the necessary context and background to place our findings in a broader scientific perspective. MF: For his active participation in discussing the clinical implications of the case, and for his insightful suggestions that helped refine the manuscript. SA: For his dedication to the revision process, meticulous proofreading, and valuable editorial feedback, which contributed significantly to the clarity and coherence of the final report. HU: For his administrative support and coordination, as well as his assistance in ensuring the ethical compliance of this study. All the authors proof read the case and approved it for submission. Additionally, we acknowledge the support of our respective institutions, which provided the necessary resources for this study.
References
- Burt MJ, George DK, Powell LW et al. (1996). Haemochromatosis—a clinical update. Med J 164(6):348-51.
Publisher | Google Scholor - Kim MK, Lim DJ, Baek KH et al. (2008). A case of transfusion-associated hemochromatosis involving multiple organs. Korean J Med. 1;75(6):709-13.
| Google Scholor - Schafer AI, Cheron RG, Dluhy R et al. (1981). Clinical consequences of acquired transfusional iron overload in adults. N Engl J Med. 5;304(6):319-24.
Publisher | Google Scholor - Allen KJ, Gurrin LC, Constantine CC et al. (2008). Iron-overload–related disease in HFE hereditary hemochromatosis. N Engl J Med. Jan 17;358(3):221-30.
Publisher | Google Scholor - Beard JL, Dawson H, Piñero DJ et al. (1996). Iron metabolism: a comprehensive review. Nutr Rev. 1;54(10):295-317.
Publisher | Google Scholor - Hahn JU, Steiner M, Bochnig S et al. (2006). Evaluation of a diagnostic algorithm for hereditary hemochromatosis in 3,500 patients with diabetes. Diabetes Care. 29(2):464-6.
Publisher | Google Scholor - Adams PC, Deugnier Y, Moirand R et al. (1997 ). The relationship between iron overload, clinical symptoms, and age in 410 patients with genetic hemochromatosis. Hepatology. 25(1):162-6.
Publisher | Google Scholor - Rivers J, Garrahy P, Robinson W et al. (1987). Reversible cardiac dysfunction in hemochromatosis. Am Heart J.113(1):216-7.
Publisher | Google Scholor - Fujisawa I, Morikawa M, Nakano Y et al. (1988). Hemochromatosis of the pituitary gland: MR imaging. Radiology. 168(1):213-4.
Publisher | Google Scholor - Himoto Y, Kanzaki S, Nomura H et al. (1995). Hypothyroidism and hypoparathyroidism in an 11 year old boy with hemochromatosis secondary to aplastic anemia. Pediatr Int. 37(4):534-6.
Publisher | Google Scholor - Shirota T, Shinoda T, Aizawa T et al. (1992). Primary hypothyroidism and multiple endocrine failure in association with hemochromatosis in a long-term hemodialysis patient. Clin Nephrol. 1;38(2):105-9.
Publisher | Google Scholor - Kwon HJ, Joo SW, Kook JH et al. (2001). Endocrinopathy in hemochromatosis patients multi-transfused for aplastic anemia. Korean J Pediatr Hematol-Oncol. 181-8.
Publisher | Google Scholor - Chern JP, Lin KH et al. (2002). Hypoparathyroidism in transfusion-dependent patients with β-Thalassemia. J Pediatr Hematol Oncol. May 1;24(4):291-3.
Publisher | Google Scholor - Bell H, Rostad B, Raknerud N et al. (1994). Computer tomography in the detection of hemochromatosis. Tidsskr Nor Laegeforen. 1;114(15):1697-9.
Publisher | Google Scholor - Howard JM, Ghent CN, Carey LS et al. (1983). Diagnostic efficacy of hepatic computed tomography in the detection of body iron overload. Gastroenterology. 1;84(2):209-15.
Publisher | Google Scholor - Guyader D, Gandon Y, Deugnier Y et al. (1989). Evaluation of computed tomography in the assessment of liver iron overload: a study of 46 cases of idiopathic hemochromatosis. Gastroenterology. 1;97(3):737-43.
Publisher | Google Scholor - Alustiza JM, Artetxe J, Castiella A et al. (2004). MR quantification of hepatic iron concentration. Radiology. 230(2):479-84.
Publisher | Google Scholor













