

case report
Elevated IgE Levels in A Patient with Myelodysplastic Syndrome
1Department of Internal Medicine, Cooper University Hospital, Camden, New Jersey, United States.
2Department of Hematology and Oncology, MD Anderson Cancer Center at Cooper, Camden, New Jersey, United States.
3Department of Pathology, Cooper University Hospital, Camden, New Jersey, United States.
*Corresponding Author: Vede Ramdass,Department of Internal Medicine, Cooper University Hospital, Camden, New Jersey, United States
Citation: Ramdass V, Tulin B. Alpdogan, Behling.E. (2023). Elevated IgE levels in a patient with Myelodysplastic Syndrome, International Journal of Medical Case Reports and Reviews, BioRes Scientia Publishers. 2(4):1-5. DOI: 10.59657/2837-8172.brs.23.030
Copyright: © 2023 Vede Ramdass, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: July 31, 2023 | Accepted: August 14, 2023 | Published: August 18, 2023
Abstract
The association between immune disorders and bone marrow failure syndromes has been noted in several congenital disorders, however for adult patients ‘concurrent presentation of an immune disorder with Myelodysplastic Syndrome has rarely been noted. In this paper, we review the clinical course of our patient who presented with elevated immunoglobulin E and Myelodysplastic syndrome. His counts improved with anti-immunoglobulin E treatment over a year, but then developed acute leukemia transformation after becoming resistant to treatment with Omalizumab.
Keywords: ige; mds; inflammatory; autoimmune
Introduction
Myelodysplastic Syndromes (MDS) are a heterogenous group of clonal stem cell disorders that present with cytopenia’s and morphologically dysplastic hematopoiesis [1,2]. The clinical manifestations of MDS are predominantly related to the bone marrow failure and subsequent cytopenia, but Autoimmune and Inflammatory conditions (AIC) are reported in patients with MDS [2]. The association between MDS and AIC is unclear, but approximately 10-28% of MDS patients are noted to have AICs. AICs influence on both MDS management and outcome are an area of ongoing research [2,3]. The most prevalent AICs that have been reported include Hypothyroidism, Idiopathic Thrombocytopenic Purpura, Rheumatoid Arthritis and Vasculitis. In this case report, we present a patient with MDS who had elevated IgE Levels at presentation, and then transformed into AML after losing response to anti-IgE treatment.
Case presentation
Patient with a Past Medical History of Asthma presented with pancytopenia and in the setting of tenosynovitis. His initial pancytopenia evaluation was unremarkable for infectious, nutritional and medication-induced causes. Despite completion of antibiotic treatment, his pancytopenia did not improve for 6 weeks, and due to patient’s persistent pancytopenia a bone marrow biopsy was performed. This initial bone marrow revealed normal hypocellular bone marrow with mild increase in blasts (Figure 2A, B). Bone marrow cytogenetic analysis revealed normal 46 XY karyotype, FISH panel was negative for chromosomal changes in 5q, 7q, 8q and 20q. According to the International Prognostic Scoring System (IPSS-R) he had intermediate risk MDS [4,6]. However, Next Gene Myeloid Panel was remarkable for four clinically significant gene variants: RUNX1, SETBP1, SRSF2 and STAG [5,7]. These genetic alterations are suggested to determine high risk for AML transformation in the proposed MIPSS-R scoring, a risk stratification system which was developed by integrating IPSS-R and the mutation scores [6,8]. As the MIPSS-R scoring has not been standardized for determining management in MDS, he was managed with supportive care (i.e., transfusions, growth factor support). The blood counts stabilized with resolution of infection, however over the next six months patient’s clinical course got complicated with reoccurrence of both neutropenia and thrombocytopenia, along with folliculitis decalvans, lymphadenopathy and frequent hospitalizations for asthma exacerbation. Given the constellation of these findings, patient was evaluated for immunodeficiency and noted to have extremely elevated IgE level (>5000 IU/mL). Interestingly, patient was never noted to have eosinophilia (during patient’s clinical course the Eosinophil Absolute Count ranged from 0 to 0.19 x 103 cells/mL with normal range 0.05-0.5 cells/mL [9]. His bone marrow DNA was tested for STAT3 mutations that are commonly associated with elevated IgE, but no variants were detected [10]. Patient was then started on omalizumab, a humanized IgG1k monoclonal antibody that specifically binds free human IgE in both blood and interstitial fluid, and to the membrane bound form of IgE on B cells [11]. With treatment, cutaneous lesions improved, along with normalization of both IgE levels and cytopenia’s (Figure 1 demonstrates improvement in both platelet counts and Absolute Neutrophil Counts (ANC) with decreasing IgE levels). His blood counts, IgE levels and folliculitis decalvans were controlled with omalizumab over 12 months, but management required adjustment of dose and frequency due to fluctuations. However, he was noted to have increasing IgE levels despite the highest dose omalizumab (375 mg every 2 weeks) along with worsening pancytopenia. A repeat bone marrow biopsy was then performed which showed 37% blasts and a diagnosis of AML from an underlying myelodysplastic syndrome was made (Figure 2C). Interestingly, both karyotyping and FISH were remarkable for 20q deletion, but no MPL, NPM1, CEBPA, C-KIT, FLT3 and IDH mutations were identified.
Patient was then started on induction therapy with cytarabine and doxyrubicin but had refractory disease that was then treated with FLAG chemotherapy regimen (fludarabine, cytarabine and filgrastim). A repeat bone marrow biopsy continued to show residual disease as shown in Figure 2D, but patient’s clinical course deteriorated with pulmonary and oral infections and acute renal failure. Due to recurrent disease and acute illness the patient was determined to have poor prognosis and decided to go on hospice.
Discussion
In the US, MDS occurs in approximately 3 to 4 persons per 105 and its frequency increases with age [12]. The clinical presentation includes cytopenia’s and their subsequent complications such as anemia or infections, autoimmune diseases and dermatological manifestations. As aforementioned, MDS is associated with some AIC such as: hypothyroidism, Idiopathic thrombocytopenic purpura, rheumatoid arthritis and other diseases that are less characterized. The pathophysiology of this process is unclear but it is suspected that the immune dysregulation in MDS can result in autoimmunity and inflammatory states [2]. In this case report we present a patient who was noted to have sustained, extremely high IgE levels when he was diagnosed with intermediate risk MDS. An elevated IgE levels- a rare phenomenon that can present with a wide range of abnormalities encompassing dermatological infections, sinopulmonary infections, and other clinical manifestations [13].
The exact pathophysiology of elevated IgE levels is unclear and it is associated with multiple genetic mutations with the most common being the STAT3 mutation [14]. Our patient was negative for STAT3; however, it is possible that another mutation may have resulted in elevated IgE [5]. In patients with elevated IgE levels who manifest with asthma, omalizumab has been used for controlling asthma symptoms [15]. The Omalizumab dose gets adjusted according to the patient’s pretreatment IgE levels, and the highest dose is 375 mg every 2 weeks if IgE levels are between 600-700 Units/ml. For our patient finding a dose was difficult, as his initial IgE level was above 10,000 Units/ml and despite treatment his levels continued to be above 1000 Unit/ml. Though his thrombocytopenia, neutropenia, clinical asthma and folliculitis decalvans symptoms improved with Omalizumab treatment, his IgE levels never decreased to normal range (Figure 1). We consider the clone that was contributing to elevated IgE levels to be never totally ablated or suppressed during his treatment. In about 12 months, he started to have increased IgE levels in spite of up titration of omalizumab and we considered that our patient’s MDS became refractory to anti-IgE treatment. The exact mechanism of resistance is unclear but there is limited evidence that Omalizumab induces antibody response. As a result, it is probable that he developed antibodies against Omalizumab but at the time there were no available testing for antibodies against Omalizumab. However, with decorating cytopenia, increasing IgE levels and resistance to Omalizumab - a repeat bone marrow was performed and diagnosed AML (see Figure 2C). Interestingly, during his AML treatment the IgE levels were suppressed, and this lends credence that the association between IgE levels and MDS is likely related to the myeloid clone. That is, as the patient progressed from MDS to AML the myeloid clone likely developed immunoreactive strategies to omalizumab and this process correlates with the increasing IgE levels that were measured. Though we were not able to show a specific gene alteration/ mutation in the STAT3 pathway, our patient’s clinic presentation was rather similar to Job’s Syndrome (Hyper-IgE syndrome) which predominantly occurs in the pediatric population and is characterized by asthma and folliculitis decalvans. However, his presentation was unique because he was an adult with no prior history of recurrent infections, and had no eosinophilia during his clinical course. It should be noted that we were not able to get any pertinent family history because he was adopted and his only child was estranged. We considered a unique case, in which a patient had elevated IgE levels, with prominent cytopenia but without eosinophilia. Suppressing the IgE level, helped his MDS clone to be controlled, but he eventually developed resistance to anti-IgE treatment. Furthermore, when cytotoxic treatments suppressed the AML clone, his IgE level was also decreased. Overall, we suggest there was a pathophysiological link between MDS/AML and this unique AIC of elevated IgE. We consider that future studies evaluate the association between MDS and AIC, and will hopefully help us develop further treatment options for MDS and improve patient outcomes [3].
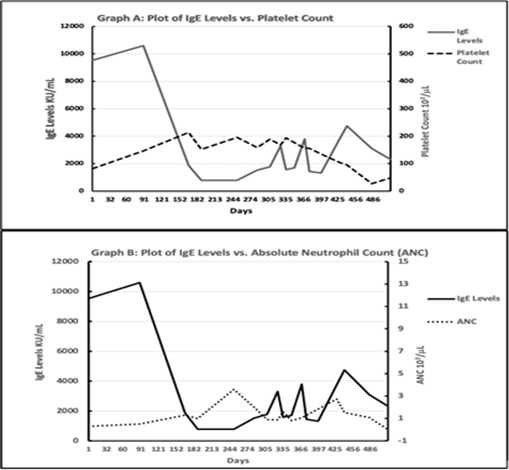
Figure 1: Graph A, B Plot of IgE levels compared to both Platelet Count & Absolute Neutrophil Count, Bone Marrow Aspiration Cytology.
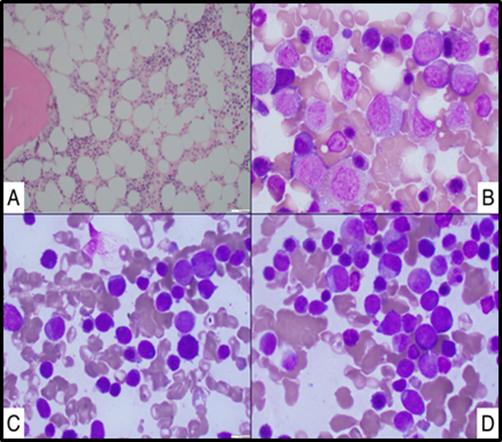
Figure 2: Graph A, B Bone Marrow Aspiration Cytology showing sightly hypocellular bone marrow with markedly left shifted myelopoiesis and mild increase in blasts. B Bone Marrow Aspiration Cytology demonstrating few blasts. C Bone Marrow Aspiration Cytology demonstrating an increase in blast replacing normal bone marrow cellularity. D Bone Marrow Aspiration Cytology demonstrating persistence of blast despite after initial AML treatment.
References
- Komrokji, R.S. et al. (2016). Autoimmune diseases and myelodysplastic syndromes. American journal of hematology, 91(5):280-283.
Publisher | Google Scholor - Wolach, O. and R. Stone. (2016). Autoimmunity and inflammation in myelodysplastic syndromes. Acta haematologica, 136(2):108-117.
Publisher | Google Scholor - Seguier, J., et al. (2019). Autoimmune diseases in myelodysplastic syndrome favors patients’ survival: a case control study and literature review. Autoimmunity reviews, 18(1):36-42.
Publisher | Google Scholor - Ogawa, S. (2019). Genetics of MDS. Blood, 133(10):1049-1059.
Publisher | Google Scholor - Al‐Shaikhly, T. and H.D. Ochs. (2019). Hyper IgE syndromes: clinical and molecular characteristics. Immunology and cell biology, 97(4):368-379.
Publisher | Google Scholor - Voso, M.T., et al. (2013). Revised International Prognostic Scoring System (IPSS) predicts survival and leukemic evolution of myelodysplastic syndromes significantly better than IPSS and WHO Prognostic Scoring System: validation by the Gruppo Romano Mielodisplasie Italian Regional Database. J Clin Oncol, 31(21):2671-2677.
Publisher | Google Scholor - Pellagatti, A. and J. Boultwood. (2015). The molecular pathogenesis of the myelodysplastic syndromes. European journal of haematology, 95(1):3-15.
Publisher | Google Scholor - Gu, S., et al. (2021). A novel scoring system integrating molecular abnormalities with IPSS-R can improve the risk stratification in patients with MDS. BMC cancer, 21(1):1-9.
Publisher | Google Scholor - Vedel-Krogh, S. (2020). The search for the “healthy” blood eosinophil count. Eur Respiratory Soc.
Publisher | Google Scholor - Heimall, J., A. Freeman, and S.M. Holland. (2010). Pathogenesis of hyper IgE syndrome. Clinical reviews in allergy & immunology, 38(1):32-38.
Publisher | Google Scholor - Chang, T.W., et al. (2007). Anti‐IgE antibodies for the treatment of IgE‐mediated allergic diseases. Advances in immunology, 93:63-119.
Publisher | Google Scholor - Montalban‐Bravo, G. and G. (2018). Garcia‐Manero, Myelodysplastic syndromes: 2018 update on diagnosis, risk‐stratification and management. American journal of hematology, 93(1):129-147.
Publisher | Google Scholor - Gernez, Y., et al. (2018). Autosomal dominant hyper-IgE syndrome in the USIDNET registry. The Journal of Allergy and Clinical Immunology: In Practice, 6(3):996-1001.
Publisher | Google Scholor - Farmand, S. and M. Sundin. (2015). Hyper-IgE syndromes: recent advances in pathogenesis, diagnostics and clinical care. Current opinion in hematology, 22(1):12-22.
Publisher | Google Scholor - Chularojanamontri, L., et al. (2009). Role of Omalizumab in a Patient with Hyper-IgE Syndrome and Review Der-matologic Manifestations. Asian Pacific Journal of Allergy and Immunology, 27(4):233.
Publisher | Google Scholor













