

Research Article
Dysimmune Encephalitis: Clinical Study
- Si Ahmed Hakim *
- Daoudi Smail
Neurology department, University Hospitalof Tizi-Ouzou, Algeria.
*Corresponding Author: Si Ahmed Hakim, Neurology department, University Hospital of Tizi-Ouzou, Algeria.
Citation: Hakim S.A, Smail D. (2023). Dysimmune Encephalitis: Clinical Study. Journal of Neuroscience and Neurological Research, BRS Publishers. 2(1); DOI: 10.59657/2837-4843.brs.23.008
Copyright: © 2023 Si Ahmed Hakim, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: January 05, 2023 | Accepted: March 30, 2023 | Published: April 04, 2023
Abstract
Introduction: Autoimmune encephalitis is a new and little-known entity targeting CNS antigens. It often causes inflammatory limbic encephalitis, for which immunotherapy is effective.
Objective: The main objective of the study is to determine the clinical and paraclinical characteristics of encephalitides associated with NMDAr, CASPR2 and LGI1 antibodies.
Materials and methods: It are a descriptive, prospective and retrospective study, which started from January 2016 to December 2018.
Results: Our cohort included 30 patients with a mean age of 44±18.05 years. The limbic encephalitis was acute in 73.3%. The patients presented mainly psychiatric and vigilance disorders, epileptic seizures, dysautonomia, and memory disorders. A temporal hyper signal (± extra limbic) was often found on MRI, with non-specific abnormalities in the EEG and an inflammatory CSF profile. FBDS, dyskinesias, and neuro myotonia were the discriminating signs. Neoplasia was found in 16.7% of the cases. Immunotherapy led to good results.
Conclusion: This study aimed to know how to identify these entities in our country and establish an early and effective treatment for better outcomes. A study with large samples would be more beneficial.
Keywords: limbic encephalitis; anti-NMDA; anti-CASPR2; anti-LGI1
Introduction
Autoimmune or dysimmune encephalitis is a rare entity, and its precise incidence and prevalence are unknown [4]. Auto antibodies bind antigens in the neuronal cell surface of the central nervous system (CNS) and unlike extra cellular antibodies, they are often responsible for inflammatory neurological damage [1, 28]. They can be seen at any age and affect both sexes, however, the age of onset and the sex distribution differ according to the type of the implicated antibody [28].
The most frequent are encephalitis with anti-NMDAr (N-methyl-D-aspartate receptors) and anti-VGKC (Voltage-Gated Potassium Channel) antibodies, the antigenic targets of which are the CASPR2 (Contact in-associated protein-like 2) and LGI1 (Leucine-rich Glioma-Inactivated 1) [18, 23]. Their clinical presentation is better individualized and often stereotyped [7,14,22,26].
The clinical manifestations are heterogeneous and depend on the type of antibody involved, even though the most common clinical presentation is an acute or subacute limbic encephalitis, which manifests with neuropsychiatric symptoms, and sometimes extra limbic manifestations, such as signs of dysautonomia, abnormal movements, dyskinesias, and ataxia [4, 16]. Some clinical signs make it possible to evoke the diagnosis and guide the search for the type of antibody involved (Faciobrachial dystonic seizures (FBDS), dyskinesias, neuromyotonic...etc.).
Their diagnosisis supported by laboratory and radiological examinations, such as the presence of inflammatory lesions on cerebral magnetic resonance imaging (MRI), an immune reaction in the cerebrospinal fluid (CSF) and an electrical pattern on the electroencephalogram (EEG). Nevertheless, it is confirmed by the detection of antibodies in the blood and/or the CSF [3,37].
Their management requires early and adapted therapeutic protocols, sometimes aggressive, involving immunomodulatory or immunosuppressive treatments [16, 50]. The response to immunotherapy is most often favourable [16, 48]. Some of these inflammatory autoimmune encephalitides can associate with tumours, and their removal is essential for better clinical outcomes [17].
We conducted a descriptive, longitudinal, prospective and retrospective study, that included thirty patients who presented a dysimmune encephalitis with antibodies against neuronal cell surface proteins (NMDAr, LGI1 and CAPSR2), within the Neurology department of the University Hospital of Tizi-Ouzou, during the period from January 2016 to December2018.
This work aimed to depict the clinical and paraclinical features of patients presenting autoimmune encephalitis associated with antibodies against extracellular epitopes of neuronal cell-surface (anti NMDAr, anti LGI1, anti CASPR2), and to assess their therapeutic response.
Patients and Methods
It is a multivariate descriptive, longitudinal, prospective and retrospective study about patients who presented dysimmune encephalitis (to anti-NMDAr, anti-LGI1 and anti-CAPSR2) in the neurology department of the Tizi-Ouzou university hospital from January 2016 to December 2018. Our study included a total of thirty patients [30].
We included patients who presented anti-NMDAr and anti-VGKC dysimmune encephalitis (CASPR2, LGI1). They have presented limbic encephalitis associating, to varying degrees, neuropsychiatric symptoms. The signs onset was either acute or subacute (in several weeks or months).
All the study patients had suggestive paraclinical examinations, namely cerebral MRI, EEG, CSF analysis, as well as anti-NMDAr, anti CASPR2, and anti LGI1 antibodies in the CSF and/or the serum.
We included patients with confirmed diagnosis (definite diagnosis) or with seronegative forms (probable diagnosis) with suggestive clinical characteristics (dyskinesias, brachio-facial dystonias, neuromyotonia, hyponatremia and dysautonomia), as well as paraclinical results (EEG patterns, MRI abnormalities, and CSF abnormalities).
Other differential diagnoses, such as herpetic encephalitis and paraneoplastic encephalitis, have been ruled out. A psychiatric evaluation was carried out in collaboration with the psychiatrists (based on the DSM-V).
Results
From January 2016 to December 2018, we identified thirty patients with one of the three types of dysimmune encephalitis (anti-NMDAr, anti-LGI1, or CASPR2). The mean duration of follow-up was 18 months, with a minimum of 06 months due to death in one patient (3.3%) and a maximum of 30 months in two patients (6.7%). These thirty patients included 16 women and 14 men, with a slight female predominance (53.3%), with a sex ratio of 0.87.
73.3 % of patients had an acute onset, while 26.7 % had a subacute onset. Prodromes were present in one-third of the patients, mainly headache, asthenia, vomiting, and photophobia. The mode of entry was psychiatric in 66.7% of cases and neurological in 33.3% of cases dominated by seizures. Vigilance disturbances were found in 66.7% of cases, to varying degrees, and 10% of patients needed tracheal intubation. Different types of seizures were found in 83.3% of cases. Faciobrachial dystonic seizures (FBDS) were observed in 26.7 % of the patients. Dyskinesias were observed in a third of cases, dysautonomic signs in 76.7% of cases and insomnia in 53.3% of cases. Many unusual symptoms have been reported including central signs, such as mutism, catatonia, aggravation under neuroleptics, pruritus, and peripheral symptoms, namely cramps, paresthesias, and fasciculations.
However, in 30% of cases, the examination revealed objective neurological signs (cerebellar ataxia, hemiparesis, extrapyramidal signs, signs of peripheral involvement), and even neuromyotonia in 20% of cases. Besides, fever was only observed in 23.3% of cases.
MMSE score showed slight memory impairment in 86.2% of cases. Biologically, a third of the patients presented with hyponatremia. Brain MRI revealed abnormalities in 86.7% of cases.
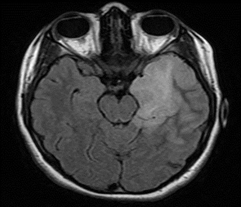
Figure 1: 17-year-old female patient with anti-NMDAr encephalitis (seropositive). Cerebral MRI, FLAIR sequence: left temporo-insular hyper signal.
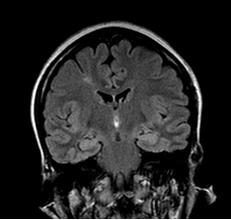
Figure 2: 65-year-old patient with anti-LGI1 encephalitis (seropositive). Cerebral MRI, FLAIR sequence: bilateral amygdalo-hippocampal hyper signal.
Cerebrospinal fluid analysis showed abnormalities in 86.7% of cases. 73.3% presented hyperproteinorachia, and only 23.3% had lymphocyte-type hypercellularity with an average of 45.86 lymphocytes / mm3. The CSF immunology was pathological in 53.35% of cases.
On the initial EEG, 76.7% of cases presented generalized or focal nonspecific cerebral abnormalities including background slowing, theta or delta activity, and only 23.3% presented with epileptic abnormalities. Furthermore, no patient presented Extreme Delta Brush, but other abnormalities were rarely observed (fast rhythms, slow rhythmic waves, and periodic complexes).
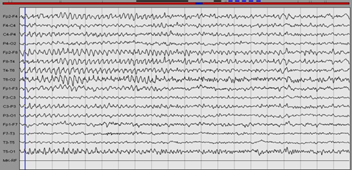
Figure 3: EEG of a patient with anti-NMDAr encephalitis: Rhythmic slow waves especially on the right.
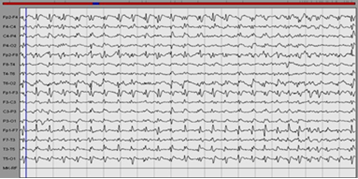
Figure 4: of a patient with anti CASPR2 encephalitis: Periodic complexes.
The specific immunological examination, which consists of measuring membrane antibodies (NMDAr, VGKC, LGI1, CSAPR2), showed that 63.3% of cases presented positive antibodies. 36.84% of patients presented anti CASPR2 antibodies, 26.32% anti-NMDAr antibodies, and 26.32% anti-LGI1 antibodies. 5.26% of cases displayed a combination of CASPR2 and anti LGI1 antibodies, and 5.26% of cases showed a combination of anti LGI1 and anti VGKC antibodies. It is worth noting that they were found in 63.2 % of cases in the CSF, 26.3% in the blood, and 10.5 % in both.
The search for associated neoplasia revealed the presence of tumour in 16.7% of cases. The mean age of the patients with the tumour was 46.6 ± 11 years. The anatomopathological study found mainly thymomas (stage B1 or B2 / B3) and adenocarcinoma of the colon.
All patients received first-line treatment, with 93.3% receiving intravenous (IV) methylprednisolone (1g/day for three to five days), 96.7% receiving IV immunoglobulin (0.4 mg/kg for 05 days) at a rate of one to six cures, and 6.7% receiving plasma exchanges at a rate of three courses). A switch to oral corticosteroids (1 mg/kg/day) was used in 30% of cases. Second-line treatment was started in 46.7% of cases. Rituximab and Cyclophosphamide were used in severe forms, while Azathioprime was initiated in the less severe cases to prevent relapses or as a relay to prior treatment in conjunction with a specific treatment of the existing tumor. One-third of patients have completely recovered, 63.3 percent have recovered significantly, and one patient has died as a result of tumor complications. Treatment improved the major symptoms (psychiatric conditions, vigilance disorders, epileptic seizures, and memory disorders) (p = 10-3). However, in 63.4 percent of cases, sequelae were observed, with memory impairment being the most common. In 80% of cases, patient autonomy was regained, with just 6.7 percent of cases experiencing a single episode of relapse.
Discussion
This work aimed to analyse the clinical and paraclinical features of autoimmune encephalitis due to antibodies targeting neuronal cell membrane antigens, namely anti-NMDAr, anti LGI1, and anti CASPR2, in order to better recognize these entities. Therefore, our findings were compared to data from the literature after an average follow-up of 18 months. In our cohort, there was a slight female predominance (sex ratio of 0.87). However, this sex-based distribution did not accurately reflect the various types of antibodies. Some types of encephalitis, such as anti-NMDAr encephalitis [28], have a significant female predominance, while others, such as anti-CAPR2 and anti-LGI1 encephalitis, have a significant male predominance. [5, 38].
In our study, the mean age was 44 ± 18 years, with a vast majority of patients over the age of 45 years. We found an autoimmune history in 26.7% of cases, favouring the disease onset, particularly in the pediatric population [12]. It was a disease that was thought to be uncommon but was likely underdiagnosed. The recent rise in the number of cases reported in the literature [4] is likely due to the availability of biological markers as well as a greater understanding of suggestive clinical presentations. Regardless of the antibody involved, the clinical presentation in our series was relatively stereotyped, with some clinical homogeneity, similar to what has been described in the literature. Limbic encephalitis is the most common presentation with mainly an acute onset (73.3 % of cases) or subacute (26.7 % of cases). It is characterized by a psychiatric (60.7%) or neurological (33.3%) entry mode in the majority of cases. The psychic manifestations include behavioural disorders, such as aggression, agitation, irritability, panic attacks and hallucinations. They are associated with neurological manifestations, such as impairment of antegrade memory (100% of cases), epileptic seizures (83.3% of cases), dysautonomia signs (76.7% of cases) and vigilance disorders (66.7% of cases) [41]. Prodromes were found after a thorough examination (33.3% of cases), although few studies were interested in this parameter, which is most commonly seen in anti-NMDAr encephalitis (70% to 86% of cases) [9].
Patients can seek treatment in psychiatry first because of the psychiatric manifestations (depression, anxiety, and psychotic manifestations), which may mislead the diagnosis [52]. On the other hand, epileptic seizures (inaugural in 23.3% of cases) dominated the neurological onset manifestations, emphasizing the importance of maintaining a critical approach in the face of a first epileptic seizure in order to avoid being satisfied with a simple antiepileptic treatment, which can be inefficient. Epileptic seizures were found in 83.3% of our patients (at the onset or in the acute phase), which was consistent with the literature [33,50]. These are mostly temporal, which is in line with previous findings [8].
Our study found vigilance disturbances ranging from simple obnubilation to coma, with 30% of our patients falling into a coma. Because of certain complications, such as infections, these disorders of consciousness may worsen the vital and functional prognosis, and may even prevent the initiation of certain immunosuppressive treatments [10].
Extra limbic symptoms have been observed in our patients, which makes the name limbic encephalitis debatable in favour of autoimmune or dysimmune encephalitis as some authors have suggested, so as not to underdiagnose certain atypical forms or purely extra limbic encephalitis [4]. As a result, Lai et al. proposed, in 2010, the term autoimmune synaptic encephalitis to replace the term limbic encephalitis [38]. Some signs and symptoms are highly suggestive of autoimmune encephalitis, and it is essential to recognize them. FBDS (26.7% of cases), dyskinesias (33% of cases), orofacial or diffuse with choreic movements and opisthotonos attitude, are considered to be the most suggestive signs. Dysautonomia signs were present in 76.7% of our patients, which approaches the data reported in the literature (69%) [10, 12]. Neuromyotonia and sometimes even paroxysmal ataxia have also been reported [28, 32, 34, 36,40]. Dysautonomia signs were one of the most defining features of these entities, which could result in cardiac and/or respiratory decompensation [10]. Many of those signs were observed in our series, namely sweating, salivation, temperature and blood pressure fluctuations. Neuromyotonia was of capital importance for the diagnosis since it was typical in certain types of antibodies (anti CASP2), allowing the search for the type of tumour that could be involved (thymoma) to be guided [34.39].
We found sleep disorders, such as insomnia, in 53.3% of cases, and it is often identified as a suggestive symptom of some dysimmune encephalitis [2, 32, 44, 49]. It is worth noting that sleep disturbances have been reported in paraneoplastic limbic encephalitis associated with anti Ma2 antibodies [11], and that was ruled out by onco-neuronal antibodies testing in our cohort. Paraclinical examinations not only rule out other differential diagnosis but they also contribute to confirm the diagnosis by pointing out certain typical characteristics.
Hyponatremia should be looked for systematically because it can worsen vigilance disturbances, and it requires appropriate correction and the use of some therapies with caution. It was found in a third of our study population. It is rarer in other aetiologies of limbic encephalitis, such as limbic paraneoplastic encephalitis (anti-Hu) compared to what has been reported in autoimmune encephalitis [20, 30, 50].
The scalp EEG was an easy, non-invasive, and cost-effective exam to perform. It was essential for the differential diagnosis (herpetic encephalitis, metabolic encephalopathies, Creutzfeld- Jacob disease). In the literature, no electrical pattern common to all antibodies has been identified. It often showed a diffuse or focal non-specific slowing (up to 90% of cases) and sometimes epileptic abnormalities [19, 31, 48]. On the other hand, an "extreme delta brush (EDB)" pattern has been described only in anti NMDAr encephalitis, which is very suggestive, and even fast rhythms or slow rhythmic waves [15, 35,47]. In our study population, the initial EEG was often pathological (76.7% of cases), often displaying the non-specific abnormalities described above [19]. Inter-critical epileptic anomalies were also uncommonly recorded. The reported EDB pattern was not found in our series, which could be explained by its transient character [19].
Brain MRI often objectified parenchymal abnormalities (86.7% of cases), which were similar to those reported in the literature [25, 51], including isolated temporal lesions (36.6% of cases). They were more bilateral than unilateral. Extra limbic radiological abnormalities were found in half of the cases. The most affected localisations are mainly the frontal lobe, the basal ganglia, and rarely other structures, such as the insula, parietal lobe, occipital lobe, meninges and white matter are affected. These extra limbic anomalies have been reported in the literature at a lower frequency than ours (40% of cases) [51]. Graus et al. included bilateral temporal involvement (in T2 and FLAIR) in the criteria for defined autoimmune encephalitis [23]. Unilateral temporal impairment would be possible. However, in this case, Graus et al. required the presence of antibodies to classify it as autoimmune encephalitis because other etiologies, such as glioma, seizures, and HSV1, could cause a unilateral temporal hyper signal [23].
The cytological and biochemical analysis of the CSF was essential, and it was primarily pathological (86.7% of our cases), which is consistent with the literature data [6,24]. CSF bacteriological analysis was essential in order to rule out infectious causes. In some series, the CSF profil was normal [38,50], which is not against the diagnosis of dysimmune encephalitis. The CSF abnormalities were mainly hyper proteinorachia (73.3% of cases, often the level did not exceed 01g/l), lymphocytic pleocytosis (23.3% of cases, with an average of 45,86 lymphocytes/mm3), and especially the presence of oligo-clonal bands (OCBs) in 46.6% of cases, which demonstrates the existence of an inflammatory mechanism, particularly for seronegative forms. These results are close to those reported by certain authors who found slight to moderate lymphocytic pleocytosis (generally less than 100 cells/mm³) in 60 to 80% of patients and oligoclonal bands in approximately 50% of cases [24, 27].
The specific antibodies testing confirmed the diagnosis, but the determination of the type of antibody to look for and the interpretation of the results had to be chosen according to the clinical presentation and the results of the paraclinical examinations and required a specialized centre with adapted techniques. Some authors have suggested that the presence of antibodies targeting CNS cell membrane was not necessary for the diagnosis of autoimmune encephalitis and that their negativity should in no way delay the initiation of an early treatment if the clinical manifestations and the para-clinical data are suggestive [4, 21]. This was the case in our study, where one-third of the patients had seronegative encephalitis and responded well to immunotherapy.
In our cohort, 63.3% of cases were seropositive (indirect immunofluorescence, immunohistochemistry, and a specific test on transfected HEK cells). Other tests were also necessary, such as the assay of onco-neuronal antibodies (antibodies targeting intracellular antigens) to rule out paraneoplastic encephalitis. We have diagnosed a significant number of patients with this latter (anti-Yo, Ma2, Ri, anti Sox1, CV2/CRMP5, anti-Tr). We also assayed the antibodies directed against intracellular synaptic antigens, which made it possible to diagnose one case of anti-GAD65 encephalitis and one case of anti-amphiphysin encephalitis.
Further exams were performed to look for associated neoplasia, namely abdominal, pelvic and thoracic CT-Scan, pelvic and scrotal ultrasound, as well as tumour markers and PSA assays. Therefore, we found a paraneoplastic syndrome in some patients (16.7% of cases) associated with thymoma or adenocarcinoma confirming the possibility of the existence of paraneoplastic encephalitis among these cases without them being purely autoimmune [1].
In terms of management, we proposed some treatment plans based on prior experiences because there were no therapeutic consensus or well-codified protocols. There was no data from randomized controlled clinical trials of immunotherapy use, the order in which they should be administered or the duration of treatment [10,13, 29,45]. Therefore, we used IV bolus of methylprednisolone as first-line treatment, which was typically followed by oral corticosteroids relay and IV immunoglobulins (Ig) [13, 29, 45]. Some experts defended plasma exchanges, which we used in just 6.7% of cases, while others prefered IV Ig [43,46], despite the lack of compelling evidence of superiority [45]. IV Ig seemed to be more manageable than plasma exchanges due to frequent agitation, epileptic seizures interfering with plasmapheresis progress, and dysautonomia signs which could be worsened by the latter [12,50]. The molecules chosen were rituximab (375mg/m2) and cyclophosphamide (750mg/m2 IV) and have been given shortly after first-line treatment to avoid having a therapeutic window [45]. In patients with less serious manifestations, azathioprine (1mg/Kg/d) was also used to minimize the risk of relapse. This second-line therapy has been implemented in 46.7% of our patients, with no significant side effects recorded. Rituximab, a monoclonal anti-CD20 antibody, is becoming more widely used in the treatment of autoimmune diseases [42]. In parallel, tumour specific management was started with either ablation of the tumour (60% of cases) or by chemotherapy combined with radiotherapy (40% of cases). Depending on the seriousness of the clinical presentation and the therapeutic response, a second-line treatment was added. This proved the importance given to this entity that is sometimes paraneoplastic, allowing us to treat tumours at an early stage.
Finally, after an average of 18 months of follow-up (a minimum of 06 months and a maximum of 30 months), we evaluated the treatment response. One patient was lost to follow-up after 08 months. We found that 33.3% of patients recovered completely, while 63% recovered partially but significantly. Just one patient died as a result of tumour complications. According to the literature data, autoimmune encephalitis is a potentially treatable condition that may be fatal. Adequate, prompt, and sometimes aggressive treatment is essential because it often results in a significant improvement or complete recovery [28]. During the study period, we noticed a few non-disabling sequelae (66.7% of cases). Therapeutic efficacy was reflected by the patient's autonomy, which was assessed by questioning him and those around him about his daily activities without using a specific rating scale. In our sample, 80% of patients regained their autonomy.
Conclusion
This study aimed to identify the clinical and paraclinical characteristics of the most common dysimmune encephalitis types: anti-NMDAr, anti-LGI1 and anti-CASPR2. For a long time, it was confused with other limbic encephalitis causes. The majority of the previous studies described the different subtypes of dysimmune encephalitis without grouping them into a single clinical and paraclinical entity to make clinical diagnosis easier without waiting for specialized examinations. This study proposed a rapid diagnostic approach based on some discriminative clinical features, namely FBDS, dyskinesias, typical psychiatric manifestations and peripheral signs. These characteristics differ depending on which antibody is involved, and the diagnosis is supported by suggestive paraclinical findings such as a temporal hyper signal, which is usually bilateral and often associated with extra limbic lesions on brain MRI, oligo-clonal bands and EEG abnormalities.
References
- Antoine. J.C. (2018). Les encéphalites auto-immunes avec auto-anticorps. Tableau clinique et stratégie diagnostique et thérapeutique. Pratique Neurologique- FMC, 9:81-87.
Publisher | Google Scholor - Arnaud. I, Mackowiak. MA, Enderle. A, Pasquier. F. (2015). Encéphalite limbique à anticorps anti-Lgi1. J. Neurol, 01.276.
Publisher | Google Scholor - Arnould. V, Dufournet. B, Boucraut. J, Boyer. L, Bou Ali. H, Gaillard Bigot. F, Pelletier. J, Mundler. O, Kaphan. E, Guedj. E. Corrélations des profils TEP au 18F-FDG, IRM et clinico-biologiques des encéphalites dysimmunitaires. Médecine Nucléaire, (41):418-425.
Publisher | Google Scholor - Aupy. J, Collongues. N, Blanc. F, Tranchant. C, Hirsch. E, De Seze. J. (2013). Autoimmune encephalitis, clinical, radiological and immunological data. Rev Neurol, 169:142-153.
Publisher | Google Scholor - Bastiaansen. AEM, Van Sonderen. A, Titulaer. MJ. (2017). Autoimmune encephalitis with anti-leucine-rich glioma- inactivated 1 or anti-contactin-associated protein-like 2 antibodies (formerly called voltage-gated potassium channel-complex antibodies). Curr Opin Neurol, 30:302-309.
Publisher | Google Scholor - Bataller. L, Kleopa. KA, Wu. GF, Rossi. JE, Rosenfeld. MR, Dalmau. J. (2007). Auto-immune limbic encephalitis in 39 patients: Immunophenotypes and outcomes. J. Neurol Neurosurg Psychiatry, 78(4):381-385.
Publisher | Google Scholor - Beaudonnet. F, Garrec. N, Sfez. A, Arnaud, Demersay. C. (2012). L’encéphalite à anticorps anti-récepteur au NMDA: Une cause sous-estimée de psychose aiguë chez l’enfant et l’adulte jeune. Adolescence & Médecine. Juillet, 41(3, P1):318-320.
Publisher | Google Scholor - Bien. CG, Elger. CE. (2007). Limbic encephalitis: A cause of temporal lobe epilepsy with onset in adult life. Epilepsy Behav, 10:529-538.
Publisher | Google Scholor - Congcong. Ma, Chengze. W, Qiaoman. Z, Yajun. L. (2019). Emerging role of prodromal headache in patients with anti- N-methyl-D-aspartate receptor encephalitis. Journal of Pain Research, 12:519-526.
Publisher | Google Scholor - Dalmau. J, Gleichman. AJ, Hughes. EG, Rossi. JE, Peng. X, Lai. M, et al. (2008). Anti NMDA-receptor encephalitis: Case series and analysis of the effects of antibodies. Lancet Neurol. Décembre, 7(12):1091-1098.
Publisher | Google Scholor - Dalmau. J, Graus. F, Villarejo. A et al. (2004). Clinical analysis of anti-Ma2-associated encephalitis. Brain. 127:1831-1844.
Publisher | Google Scholor - Dalmau. J, Lancaster. E, Martinez-Hernandez. E, Rosenfeld. MR, Balice-Gordon. R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol, 10(1):63-74.
Publisher | Google Scholor - De Broucker. T. (2015). Le Top des publications sur les encéphalites auto-immunes. Neurologies. Janvier, 18:174.
Publisher | Google Scholor - De Broucker. T, Martinez-Almoyna. L. (2010). Encéphalites avec anticorps anti-récepteur NMDA. J. Médecine et maladies infectieuses, (40):1-5.
Publisher | Google Scholor - Di Capua. D, García-Ptacek. S, García-García. ME, Abarrategui. B, Porta Etessam. J, García-Morales. I. (2013). Extreme delta brush in a patient with anti-NMDAR encephalitis. Epileptic Disord Int Epilepsy J Videotape, 15(4):461-464.
Publisher | Google Scholor - Didelot. A, Honnorat. J. (2009). Les encéphalites limbiques. La Lettre du Neurologue, 13(11).
Publisher | Google Scholor - Eté. M, Cally. R, Patient. M, Poullin. P, Boucraut. J, Bernit. E, Ebbo. M, Harlé. J.R, Kaphan. E, Schleinitz. N. (2015). Amélioration spectaculaire d’une encéphalite limbique à anti-contactin-associated protein-like 2 (CASPR2) par des échanges plasmatiques. La Revue de médecine interne, (36S):A100-A211.
Publisher | Google Scholor - Faivre. G, Klapczynski. F, Ghanim. Z, Benmerad. M, Ameri. A. (2011). Découverte d’une encéphalite limbique à anticorps anti-VGKC lors d’une consultation mémoire. Pratique Neurologique. FMC, 2:28-33.
Publisher | Google Scholor - Gagnol. A, De Montaudouin. M. Description des caractéristiques EEG d’une série de 6 patients diagnostiqués pour une encéphalite dysimmunitaire. J.Neucli.
Publisher | Google Scholor - Geschwind. M. D, Tan. K. M, Lennon. V. A, Barajas. R. F, Haman. A, Klein. C. J, Josephson. S. A, Pittock. S. J. (2008). Voltage-Gated Potassium Channel Autoimmunity Mimicking Creutzfeldt-Jakob Disease. Arch Neurol, 65(10):1341-1346.
Publisher | Google Scholor - Goetz. J, Olsson. N. O, Humbel. R. L. (2013). Anti-corps anti neuronaux. EMC biologie médicale, 90-30-008- A.
Publisher | Google Scholor - Granerod. J, Ambrose. HE, Davies. NW, Clewley. JP, Walsh. AL, Morgan. D, et al. (2010). Causes of encephalitis and differences in their clinical presentations in England: A multicentre, population-based prospective study. Lancet Infect Dis, 10(12):835-844.
Publisher | Google Scholor - Graus. F, Titulaer. MJ, Balu. R, Benseler. S, Bien. CG, Cellucci. T, et al. (2016). A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol, 15:391-404.
Publisher | Google Scholor - Gultekin. SH, Rosenfeld. MR, Voltz. R, Eichen. J, Posner JB, Dalmau J. (2000). Paraneoplastic limbic encephalitis: Neurological symptoms, immunological findings and tumour association in 50 patients. Brain J Neurol, 123(7):1481-1494.
Publisher | Google Scholor - Heine. J, Prüss. H, Bartsch. T, Ploner. CJ, Paul. F, Finke. C. (2015). Imaging of autoimmune encephalitis relevance for clinical practice and hippocampal function. Neuroscience, 309:68-83.
Publisher | Google Scholor - Henry. C, Husson. H, De Broucker. T. (2009). Encéphalite limbique auto-immune avec anticorps antirecepteur NMDA associée à un tératome de l’ovaire: Une forme curable d’encéphalite limbique paranéoplasique. Revue Neurologique, 165:70-75.
Publisher | Google Scholor - Hoftberger. R, Titulaer. MJ, Sabater. L, Dome. B, Rozsas. A, Hegedus. B, et al. (2013). Encephalitis and GABAB receptor antibodies: novel findings in a new case series of 20 patients. Neurology, 81:1500-1506.
Publisher | Google Scholor - Honnorat. J, Joubert. B. (2018). Movement disorders in autoimmune encephalitis and paraneoplastic neurological syndromes. J.Neurol.
Publisher | Google Scholor - Hung. TY, Foo. NH, Lai. MC. (2011). Anti-N-Methyl-D-aspartate receptor encephalitis. Pediatr Neonatol, 52:361-364.
Publisher | Google Scholor - Irani. SR, Alexander. S, Waters. P, et al. (2010). Antibodies to Kv1 potassium channel-complex leucine-rich, gliomainactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and aquired neuromyotonia. Brain, 133:2734-2748.
Publisher | Google Scholor - Irani. SR, Bera. K, Waters. P, et al. (2010). N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non paraneoplastic disorder of both sexes. Brain, 133:1655-1667.
Publisher | Google Scholor - Irani. SR, Gelfand. JM, Al-Diwani. A, Vincent. A. (2014). Cell-surface central nervous system autoantibodies: clinical relevance and emerging paradigms. Ann Neurol, 76:168-184.
Publisher | Google Scholor - Irani. SR, Michell. AW, Lang. B, et al. (2011). Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol, 69:892-900.
Publisher | Google Scholor - Irani. SR, Pettingill. P, Kleopa. KA, et al. (2012). Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol, 72:241-255.
Publisher | Google Scholor - Jeannin-Mayer. S, Rosenberg. S, Convers. P, Bicilli. E, Antoine. J-.C, Mazzola. L. (2016). Analyse électroencéphalographique intercritique des encéphalites à anticorps anti-récepteurs NMDA: étude de 10 cas. J.neucli.
Publisher | Google Scholor - Joubert. B, Gobert. F, Thomas. L, Saint-Martin. M, Desestret. V, Convers. P, et al. (2017). Autoimmune episodic ataxia in patients with anti- CASPR2 antibody-associated encephalitis. Neurol Neuroimmunol Neuroinflamm, 4:e371.
Publisher | Google Scholor - Lagarde. S, Guedj. E. (2015). Apport de la tomographie par émission de positon (TEP) cérébrale dans les encéphalites dysimmunitaires. Med Nucl, 39:279-282.
Publisher | Google Scholor - Lai. M, Huijbers. MG, Lancaster. E, Graus. F, Bataller. L, Balice- Gordon. R, et al. (2010). Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol, 9:776-785.
Publisher | Google Scholor - Lancaster. E, Huijbers. MG, Bar. V, et al. (2011). Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol, 69:303-311.
Publisher | Google Scholor - Lancaster. E, Martinez-Hernandez. E, Titulaer. MJ, Boulos. M, Weaver. S, Antoine. JC, et al. (2011). Antibodies to metabotropic glutamate receptor 5 in the Ophelia syndrome. Neurology, 77:1698-1701.
Publisher | Google Scholor - Lang. P. O, Sellal. F. (2008). Une encéphalite limbique non paranéoplasique révélée par une amnésie antérograde. Presse Med, 37:775-782.
Publisher | Google Scholor - Le Moigno. L, Ternant. D, Paintaud. G, Thibault. G, Cloarec. S, Tardieu. M, Lagrue. E, Castelnau. P. (2014). Encéphalite à anticorps anti-récepteurs N-méthyl-D-aspartate (NMDA-R): place des immuno-modulateurs. J. Arcped.
Publisher | Google Scholor - Mirza. MK, Pogoriler. J, Paral. K, et al. (2011). Adjunct therapeutic plasma exchange for anti-N-methyl-D- aspartate receptor antibody encephalitis: a case report and review of litterature. J Clin Apher, 26:362-365.
Publisher | Google Scholor - Montiel. P, Sellal. F, Clerc. C, Richard. P, Bataillard. M. (2008). Encéphalite limbique avec anticorps anti canaux potassiques voltage dépendants et troubles sévères du sommeil. Revue neurologique, (164):181-184.
Publisher | Google Scholor - Newman. M, Airey. C, Blum. S, Scott. J. G, Wong. R. C, Gillis. D. (2018). Autoimmune Encephalitis: Clinical Features, Pathophysiology, and Management. Neuroinflammation.
Publisher | Google Scholor - Nunez-Enamorado. N, Camacho-Salas. A, Belda-Hofheinz. S, et al. (2012). Fast and spectacular clinical response to plasmapheresis in a paediatric case of anti-NMDA encephalitis. Rev Neurol, 54:420-424.
Publisher | Google Scholor - Schmitt. SE, et al. (2012). Extreme delta brush: a unique EEG pattern in adults with anti-NMDA receptor encephalitis. Neurology, 79:1094-1100.
Publisher | Google Scholor - Titulaer. MJ, Mc Cracken. L, Gabilondo. I, Armangué. T, Glaser. C, Iizuka. T, et al. (2013). Treatment and prognostic factors for long-term outcome in patients with anti NMDA receptor encephalitis: an observational cohort study. Lancet Neurol, 12(2):157-165.
Publisher | Google Scholor - Van Sonderen. A, Thijs. RD, Coenders. EC, et al. (2016). Anti-LGI1 encephalitis: Clinical syndrome and long-term follow-up. American Academy of Neurology.
Publisher | Google Scholor - Vincent. A, Buckley. C, Schott. JM, Baker. I, Dewar. B, Detert. N, et al. (2004). Potassium channel antibody- associated encephalopathy: A potentially immunotherapy-responsive form of limbic encephalitis. Brain, 127(3):701-712.
Publisher | Google Scholor - Wagner. J, Schoene-Bake. JC, Malter. MP, Urbach. H, Huppertz. HJ, Elger. CE, et al. (2013). Quantitative Flair analysis indicates pre-dominant affection of the amygdala in antibody-associated limbic encephalitis. Epilepsia, 54(9):1679-1687.
Publisher | Google Scholor - Zandi. MS, Irani. SR, Lang. B, Waters. P, Jones. PB, McKenna. P, et al. (2011). Disease relevant autoantibodies in first episode schizophrenia. J Neurol, 258(4):686-688.
Publisher | Google Scholor











