

Review Article
Differential Diagnosis of Diffuse Cystic Lung Disease at HRCT - An Overview
1Medical Faculty, University Clinic of Pulmonology and Allergology, Skopje, Republic of Macedonia.
2Medical Faculty, University Institute of Radiology, Skopje, Republic of Macedonia.
3Medical Faculty, State Hospital 8th September, Skopje, Republic of Macedonia.
*Corresponding Author: Kristina Dimitrijevikj, Medical Faculty, University Clinic of Pulmonology and Allergology, Skopje, Republic of Macedonia.
Citation: Dimitrijevikj K., Nikolova S., Mitreska N., Prgova-Veljanovska B. (2024). Differential Diagnosis of Diffuse Cystic Lung Disease at HRCT - An Overview, International Journal of Biomedical and Clinical Research, BioRes Scientia Publishers. 2(1):1-7. DOI: 10.59657/2997-6103.brs.24.039
Copyright: © 2024 Kristina Dimitrijevikj, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: November 22, 2024 | Accepted: December 20, 2024 | Published: December 27, 2024
Abstract
Introduction: The diffuse cystic lung diseases (DCLDs) are a diverse group of lung disorders characterized by the presence of multiple regular or irregular spherical parenchymal lucencies bordered by a thin wall and having a well-defined interface with normal lung. Other lucent lung lesions like centrilobular emphysema, cavity, cystic bronchiectasis, honeycomb cyst, and pneumatoceles are close mimics of a lung cyst on high-resolution computed tomography (HRCT) HRCT is an important modality in the evaluation of interstitial lung disease to include cystic lung disease. This review describes a simple algorithmic approach for DCLDs on HRCT based on cyst’s distribution, size, and shape, as well as background parenchymal changes and it helps also in differentiation of common and uncommon diffuse cystic lung disease.
Aim: The aim of the study is to present, describe and make differential diagnosis of this spectrum of diseases associated with air cysts at high-resolution CT.
Conclusion: Diffuse cystic lung diseases are a group of complex disorders that often have an overlapping clinical presentation, but different underlying pathological processes. HRCT still remains the imaging of choice for the diagnosis of common diffuse cystic lung diseases.
Keywords: cystic disease; lung; differential; HRCT
Introduction
The diffuse cystic lung diseases (DCLDs) are a diverse group of lung disorders characterized by the presence of multiple regular or irregular spherical parenchymal lucencies bordered by a thin wall and having a well-defined interface with normal lung [1]. On chest CT, a cyst is seen as an area with a low attenuation coefficient in the lung parenchyma, having a well-defined interface with the adjacent normal lung and usually containing air, although it can occasionally have a liquid or solid content [2]. A lung cyst can originate from various mechanisms, such as airway obstruction with distal airspace dilatation (check-valve mechanism), necrosis of the airway walls, and lung parenchymal destruction by proteases [3]. HRCT has broadened the understanding of diffuse cystic lung diseases, because it enables improved identification of cyst characteristics (including their distribution) and associated lesions (including assessment of the presence of extrapulmonary changes), narrowing the differential diagnosis and often avoiding the need for diagnostic confirmation by lung biopsy [4]. Many diffuse lung diseases may manifest cysts as the primary abnormality, although lymphangioleiomyomatosis and Langerhans cell histiocytosis (LCH) are the most common to present with diffuse lung cysts [9,10]. Air-containing lesions like cystic bronchiectasis, centrilobular emphysema, bulla, cavity, honeycomb cyst, and pneumatoceles have a significant overlap in the imaging features and are often confused with cystic lung diseases. Therefore, exclusion of such overlapping mimics is very crucial to reach a definitive diagnosis of DCLDs [13,21]. The object of this article is to illustrate and describe the spectrum of diseases associated with air cysts at high-resolution CT (HRCT).
Lung Cyst Mimics
Bronchiectasis is irreversible dilatation of the bronchial tree, and is commonly accompanied by bronchial wall thickening and mucoid impaction [14]. It is usually associated with infections, genetic disorders (cystic fibrosis) or fibrosis. On HRCT, pathological airways appear ballooned, usually exceeding 2 cm in diameter. Visualizing the branching appearance of bronchi with classical tram track and signet ring sign may help differentiate bronchiectasis from the lung cysts. The cluster of grape appearance of the airways may be seen in the region of bronchiectasis [1].
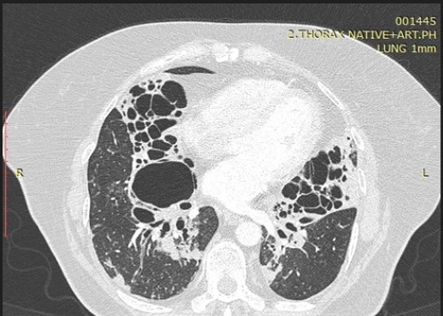
Figure 1: Inflamed cystic bronchiectasis.
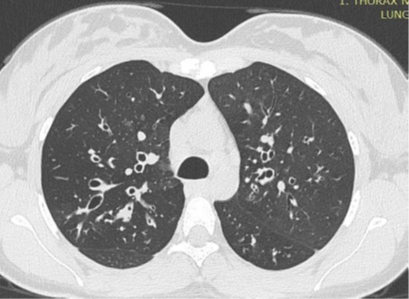
Figure 2: HRCT scan through the upper lobes of an individual with cystic fibrosis showing cylindrical bronchiectasis.
Pneumatoceles: Pneumatoceles are intrapulmonary gas-filled cystic spaces that can have a variety of sizes and appearances. The majority of pneumatoceles occur as a result of pneumonia (post-infectious pneumatocele) [15]. They are more commonly seen in children. Acute bacterial pneumonia caused by Staphylococcus aureus, Klebsiella, Pneumococcus, or Pneumocystis jirovecii sometimes produces pneumatoceles. Imaging appearance of the pneumatocele is often similar to a cyst; however, they are fewer in number and associated with adjacent consolidation or ground glass opacity as a sign of prior or resolving infection.
Pulmonary Bullae: Pulmonary bullae (singular bulla) are a gas-containing cystic structures formed by confluent destroyed and dilated airspaces (distal to terminal bronchioles). Although bullae are typically larger than blebs, there is no defining size limit for either entity in the 2024 Fleischner glossary, although a size of >1 cm was defined in the 2008 Fleischner glossary [1,14]. The most common cause is paraseptal emphysema but bullae may also be seen in association with centrilobular emphysema. They are often subpleural in location (see: subpleural bullae) and are typically larger in the apices. In some cases, bullae can be very large and result in compression of adjacent lung tissue.
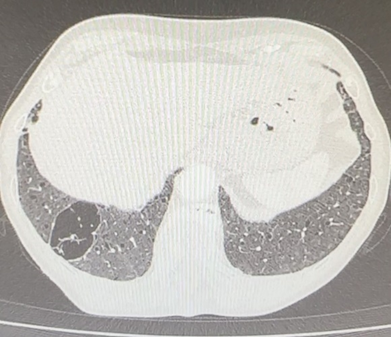
Figure 3: Subpleural septate bullae.
Emphysema: Emphysema is characterized by permanent and abnormal dilation of airways distal to the terminal bronchiole with associated destruction of their walls. On HRCT, an identifiable thin imperceptible wall in pulmonary emphysema is an essential feature that differentiates it from the cystic lung diseases. Increased lung volume and lucency, an upper lobes predominant distribution and a central dot sign representing the pulmonary artery further helps confident detection of emphysema [1].
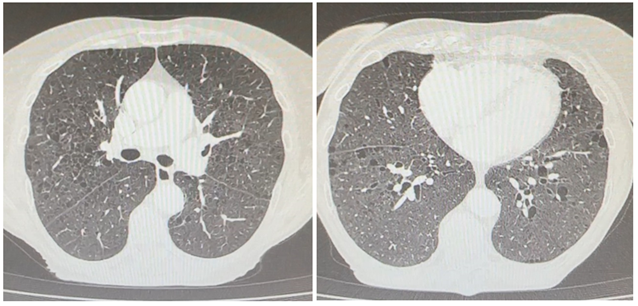
Figure 4
Cavity: Cavity is a lesion with a thick irregular wall (generally more than 4 mm) that may form within pulmonary consolidation, a mass, or a nodule. Pulmonary cavities can be congenital or secondary to infection, malignancy, previous trauma, and non-infective granulomatous pathology (as in rheumatoid nodules). The wall thickness, characteristics of its inner lining (whether smooth or irregular) and other pulmonary and extrapulmonary findings enable differentiation between the two entities [1]. Knowledge of the duration of the disease further helps to narrow the diagnosis.
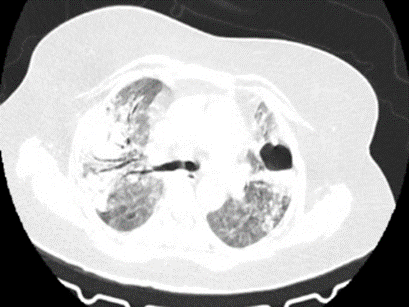
Figure 5: Cavitating lesion in the left lobe.
In honeycomb cysts, cystic changes may also be seen in fibrosing interstitial pneumonia in the form of honeycombing. Honeycombing represents the late stage of interstitial lung disease with severe fibrosis, and is characteristic of usual interstitial pneumonia. On HRCT, honeycomb cysts are characterized by well-defined cystic space of size ranging from 3 to 10 mm, having thick shared walls and a multi-layered appearance. They are clustered in the subpleural region, often with a basal predominance [1]. Associated architectural distortion, traction bronchiectasis, and volume loss can be seen. The imaging findings in combination of disease course is quite diagnostic and differentiates them from the cystic lung diseases [1].
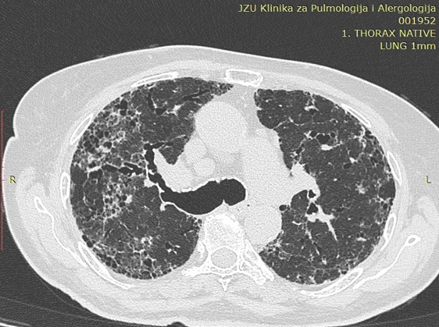
Figure 6.
Common Diffuse Cystic Lung Disease
Lymphocytic Interstitial Pneumonia: Lymphocytic interstitial pneumonia (LIP) is a benign lymphoproliferative disorder characterized by pulmonary infiltration of lymphocytes and plasma cells. It most commonly occurs in patients with Sjögren syndrome and acquired immune deficiency syndrome but can also occur in various other diseases [5,6]. The dominant HRCT finding in LIP is usually diffuse bilateral ground-glass opacity. Perivascular cysts or, less commonly, perivascular honeycombing can also be seen. Reticular pattern is seen in about half of patients. Fine nodules with a centrilobular distribution and widespread consolidation may occur. Other findings may include thickening of the bronchovascular bundles, interlobular septal thickening and lymphadenopathy [7,8].
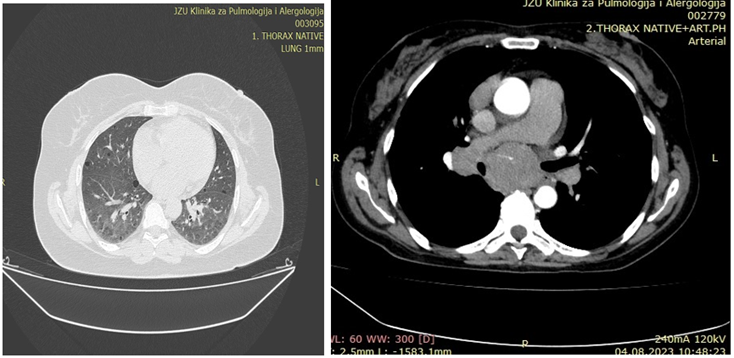
Figure 7.
Lymphangioleiomyomatosis: Lymphangioleiomyomatosis is a rare disorder occurring almost exclusively in women of childbearing age. Globally, lymphangioleio-myomatosis associated with tuberous sclerosis complex is 5- to 10-fold more common than sporadic lymphangioleiomyomatosis [10]. Characteristic HRCT features of lymphangioleiomyomatosis are diffuse thin-walled cysts surrounded by normal lung without regional sparing. Cysts are usually 2-5 mm but can be as large as 25-30 mm. Cysts are typically round or ovoid, but they may become polygonal with severe parenchymal involvement. Focal ground-glass opacities may be due to smooth-muscle cells proliferation, hemosiderosis, or pulmonary hemorrhage [11]. Obstruction of a terminal bronchiole by these abnormal cells is considered as a responsible factor for dilatation of distal air spaces and lung parenchymal destruction causing cyst formation [16].
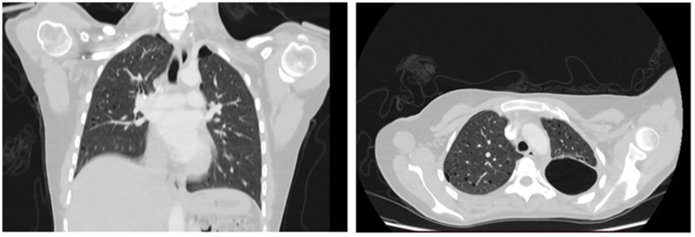
Figure 8: LAM- discrete pneumothorax on the right side, and focal liquidopneumotorax on the left. In addition, diffuse multiple thin-walled cysts were noted bilaterally and predominantly with proximal and middle zonal distribution, and no other parenchymal abnormality visible.
Pulmonary Langerhans Cell Histiocytosis: Pulmonary LCH is a smoking-related lung disease, with 80–100% of cases seen in patients who smoke or have a history of smoking. LCH occurs most frequently in young adults. On HRCT nodules may subsequently cavitate and form thick- and thin-walled cysts thought to represent enlarged airway lumina [12]. Frequently, both nodules and cysts are seen. Cysts may be round but are often irregular, bilobed, cloverleaf-shaped, or bizarre shapes. Irregular cysts, cysts with nodules, and upper zone predominance with sparing of the costophrenic angles are features that distinguish LCH from lymphangioleiomyomatosis [11].
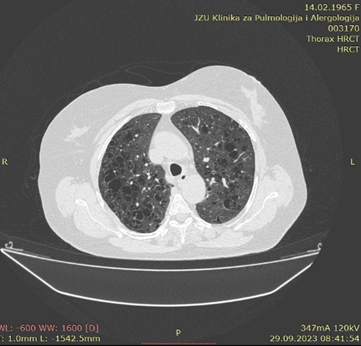
Figure 9: Pulmonary Langerhans Cell Histiocytosis.
Uncommon Diffuse Cystic Lung Disease
Amyloidosis: Although uncommon, diffuse cystic lung diseases can also develop in amyloidosis, where lung cysts are multiple (usually >10), round or lobulated, thin-walled, and variable in size [17,18].
Hypersensitivity Pneumonitis: Few scattered cysts may be seen in up to 13% of subacute and rarely in the chronic phase of hypersensitivity pneumonitis (HSP) [36]. These cysts are seen interspersed in predominant lung findings of GGOs, centrilobular nodules, areas of decreased lobular attenuation, and fibrosis. The cysts are few, uniform, and small in size, and resemble that of LIP [19].
Cystic Metastasis: Metastasis from peripheral sarcomas, angiosarcoma and mesenchymal tumors can show cystic changes and are complicated by pneumothorax. Low-grade mucosa-associated lymphoid tissue lymphomas rarely present as cystic lesions [38]. Thus, in patient with known malignancy, the appearance of a new cyst on HRCT is worrisome and requires a biopsy to rule out metastasis [20].
Practical Approach for Cystic Lung Disease
Situation 1: If we have a patient who has a cystic lesion on HRCT image, it is necessary to detect whether it is a true cyst or not. If it is not real, we should think on cavitation, cystic bronchiectasis or centrilobular emphysema as a differential diagnosis.
Situation 2: If the cyst is real, we need to determine its location, whether it is intraparenchymal or subpleural. If it is subpleural, differential diagnosis should consider bulla, paraseptal emphysema or honeycomb lung.
Situation 3: If the cyst is intraparenchymal, we should look for other additional HRCT findings. If there are no other findings, we should look for cyst distribution whether is solitary or incidental. If it is incidental, we should consider as differential diagnosis incidental cysts, pneumatocele or bronchogenic cyst.
Situation 4: If the cysts are intraparenchymal and multifocal or diffuse, we should consider on PLCH, LAM, LIP, infection, malignancy, tracheobronchial papillomatosis or BH (Birt Hogg Dube syndrome).
Situation 5: If the cysts are intraparenchymal and there are ground glass opacities as differential diagnosis we should think on DIP (Desquamative interstitial pneumonia) or PJP (Pneumocystis jirovecii pneumonia).
Situation 6: If the cysts are intraparenchymal and there are nodules as additional finding, we should consider on amyloidosis, LCDD (Light chain deposition disease), LIP or PLCH.
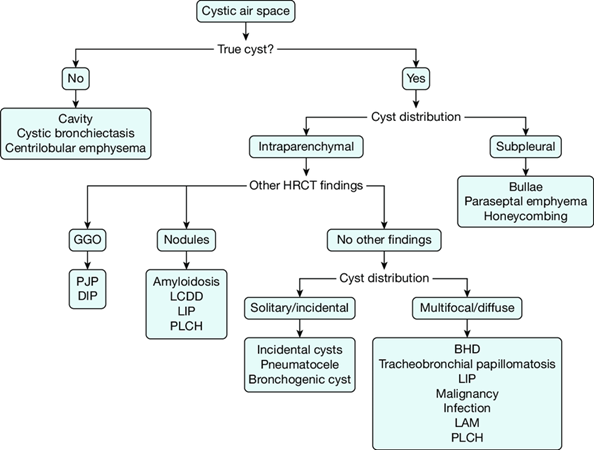
Figure 10: Kato, Takafumi & Muto, Hideharu & Hishima, Tsunekazu & Kawashima, Masahiro & Nagai, Hideaki et al. (2018). A 56-Year-Old Woman with Multiple Pulmonary Cysts and Severe Chest Pain. Chest. 153:e105-e112.
Conclusion
DCLD is an uncommon clinical and radiographic presentation with a wide spectrum of differential diagnosis. HRCT is a golden standard in the evaluation of patients with diffuse cystic lung disease. It shows us more precisely the characteristics and distribution of the cysts within the parenchyma. The differential diagnoses of diffuse cystic lung diseases are myriad, including neoplastic, inflammatory, and infectious etiologies. When combined with clinical and laboratory findings, HRCT is often sufficient for the etiological definition of diffuse lung cysts, avoiding the need for lung biopsy.
References
- Hansell DM, Bankier AA, MacMahon H, McLoud TC, Müller NL, et al. (2008). Fleischner Society: Glossary of Terms for Thoracic Imaging. Radiology. 246:697-722.
Publisher | Google Scholor - Gupta N, Vassallo R, Wikenheiser-Brokamp KA, McCormack FX. (2015). Diffuse Cystic Lung Disease Part II. Am J Respir Crit Care Med. 192(1):17-29.
Publisher | Google Scholor - Silva CI, Marchiori E, Souza AS, Júnior, Müller NL. (2010). Illustrated Brazilian Consensus of Terms and Fundamental Patterns in Chest CT Scans. J Bras Pneumol. 36(1):99-123.
Publisher | Google Scholor - Ferreira Francisco FA, Soares Souza A, Jr, Zanetti G, Marchiori E. (2015). Multiple Cystic Lung Disease. Eur Respir Rev. 24(138):552-564.
Publisher | Google Scholor - Panchabhai TS, Farver C, Highland KB. (2016). Lymphocytic Interstitial Pneumonia. Clin Chest Med. 37:463-474.
Publisher | Google Scholor - Honda O, Johkoh T, Ichikado K, et al. (1999). Differential Diagnosis of Lymphocytic Interstitial Pneumonia and Malignant Lymphoma on High-Resolution CT. Am J Roentgenol. 173:71-74.
Publisher | Google Scholor - Fischer, T., Reynolds, J., Trotter, S. (2004). The Idiopathic Interstitial Pneumonias: A Beginner's Guide. Imaging. 16:37-49.
Publisher | Google Scholor - David A. Lynch, William D. Travis, Nestor L. Muller, Jeffrey R. Galvin, David M. Hansell, et al. (2005). Idiopathic Interstitial Pneumonias: CT Features. Radiology, 236:10-21.
Publisher | Google Scholor - Abbott GF, Rosado-de-Christenson ML, Frazier AA, Franks TJ, Pugatch RD, et al. (2005). From The Archives of the AFIP: Lymphangioleiomyomatosis: Radiologic-Pathologic Correlation. Radiographics. 25(3):803-828.
Publisher | Google Scholor - McCormack FX. (2008). Lymphangioleiomyomatosis: A Clinical Update. Chest. 133(2):507-516.
Publisher | Google Scholor - Seaman DM, Meyer CA, Gilman MD, McCormack FX. (2011). Diffuse Cystic Lung Disease at High-Resolution CT. Am J Roentgenol. 196(6):1305-1311.
Publisher | Google Scholor - Abbott GF, Rosado-de-Christenson ML, Franks TJ, Frazier AA, Galvin JR. (2004). From The Archives of the AFIP: Pulmonary Langerhans Cell Histiocytosis. Radiographics. 24(3):821-841.
Publisher | Google Scholor - Singh P, Verma AK, Pandey G. (2022). Diffuse Cystic Lung Diseases: Imaging Spectrum and Diagnostic Approach Using High-Resolution Computed Tomography. Lung India. 39(6):553-561.
Publisher | Google Scholor - Bankier AA, MacMahon H, Colby T, Gevenois PA, Goo JM, et al. (2024). Fleischner Society: Glossary of Terms for Thoracic Imaging. Radiology. 310(2):e232558.
Publisher | Google Scholor - Gaillard F, Knipe H, Feger J, et al. (2024). Pneumatocele. Reference Article, Radiopaedia.
Publisher | Google Scholor - Corrin B, Liebow AA, Friedman PJ. (1975). Pulmonary Lymphangiomyomatosis. A Review. Am J Pathol. 79(2):348-382.
Publisher | Google Scholor - Baqir M, Kluka EM, Aubry MC, Hartman TE, Yi ES, et al. (2013). Amyloid-Associated Cystic Lung Disease in Primary Sjögren's Syndrome. Respir Med. 107(4):616-621.
Publisher | Google Scholor - Colombat M, Stern M, Groussard O, Droz D, Brauner M, et al. (2006). Pulmonary Cystic Disorder Related to Light Chain Deposition Disease. Am J Respir Crit Care Med. 173(7):777-780.
Publisher | Google Scholor - Boiselle PM, Crans CA Jr, Kaplan MA. (1999). The changing face of Pneumocystis carinii pneumonia in AIDS patients. Am J Roentgenol. 172(5):1301-1309.
Publisher | Google Scholor - Yogi A, Miyara T, Ogawa K, Iraha S, Matori S, et al. (2016). Pulmonary Metastases from Angiosarcoma: A Spectrum of CT Findings. Acta Radiol. 57(1):41-46.
Publisher | Google Scholor - Singh P, Verma AK, Pandey G. (2022). Diffuse Cystic Lung Diseases: Imaging Spectrum and Diagnostic Approach Using High-Resolution Computed Tomography. Lung India. 39(6):553-561.
Publisher | Google Scholor














