

Research Article
Designing, Docking, Molecular Dynamics of Several Aminocoumarin Analogues to Predict Solution of Aminocoumarin Derivatives Issues: Poor Oral Bioavailability, Low Solubility, and Toxicity
1Department of Pharmacy, Islamic University, Bangladesh.
2Department of Biotechnology & Genetic Engineering, Islamic University, Kushtia, Bangladesh.
*Corresponding Author: Md. Al-Amin, Department of Pharmacy, Islamic University, Bangladesh.
Citation: Amin A, Haque A. (2024). Designing, Docking, Molecular Dynamics of Several Aminocoumarin Analogues to Predict Solution of Aminocoumarin Derivatives Issues: Poor Oral Bioavailability, Low Solubility, and Toxicity. Scientific Research and Reports, BioRes Scientia Publishers. 1(1):1-8. DOI: 10.59657/2996-8550.brs.24.002
Copyright: © 2024 Al-Amin, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: December 20, 2023 | Accepted: January 09, 2024 | Published: January 16, 2024
Abstract
Aminocoumarin derivatives such as Novobiocin, Coumermycin, and Clorobiocin have several issues: poor oral bioavailability, low solubility, and toxicity. The Food and Drug Administration (FDA) withdrew Novobiocin sodium from the market for safety and efficacy issues in 2009. Adding selective moieties to the aminocoumarin skeleton may make it possible to design effective aminocoumarin analogues that will solve the issues of the aminocoumarin derivatives. Based on the drug design criterion, we designed several aminocoumarin analogues by adding alicyclic and aliphatic moieties at the modifiable positions of the aminocoumarin skeleton, trifluoroethylamine as bioisosteres of the amide bond of the aminocoumarin skeleton, and added fluorine to the metabolic sites of the aminocoumarin skeleton. After docking, it was understood that the designed aminocoumarin analogues effectively blocked the active site's amino acids such as Asn54, Asp81, and Arg84 of the DNA gyrase of Staphylococcus aureus.
Moreover, molecular dynamics data suggested that the designed analogue-3 had good stability compared to Novobiocin. In addition, the designed analogues-2 and 3 followed Lipinski's rule of five, which predicts that they will contain an excellent oral bioavailability profile. Furthermore, the solubility and toxicity analysis indicated that the designed analogues had good solubility and less toxicity.
Keywords: aminocoumarin; bioisosteres; fsp3; docking
Introduction
The aminocoumarin's skeleton is 3-amino-4,7-dihydroxycoumarin [1]. Examples of aminocoumarin antibiotics are (1) Novobiocin, (2) Coumermycin, (3) Clorobiocin, [2]. The aminocoumarin derivatives are produced from many Streptomyces species, such as Novobiocin from Streptomyces niveus [3]. Aminocoumarin targets the DNA gyrase of bacteria.[4] DNA gyrase is a type II topoisomerase found in eukaryotic and prokaryotic cells. It consists of two subunits: GyrA and GyrB. The two subunits form an A2B2 hetero-tetramer in prokaryotic and a homodimer in eukaryotic. During the DNA replication process, DNA gyrase produces negative supercoiling by cleaving and rejoining the two strands of replicated two DNA strands. For this catalytic activity of the DNA gyrase, it requires ATP binding [5, 6]. The N-terminal sub-domain of the DNA gyrase B is the ATP binding site that is targeted by aminocoumarin derivatives [7]. Asn54, Asp81, and Arg84 are the vital amino acids of the ATP binding site in the N-terminal domain of DNA gyrase B of Staphylococcus aureus which are selectively blocked by aminocoumarin derivatives [8]. However, the aminocoumarin derivatives have poor oral bioavailability, toxicity, and solubility issues [9]. We designed several analogues of aminocoumarin according to the drug design criterion to predict such derivatives, which may solve the aminocoumarin derivative's issues. We used the DNA gyrase of Staphylococcus aureus as a target for the designed analogues. Staphylococcus aureus causes bacteremia, endocarditis, osteoarticular, skin, soft tissue infections, etc. [10].
Methods and Materials
Designing and optimization of ligands
We sketched the 2D structure of the aminocoumarin analogues on the ChemDraw Professional 16.0 Drawer. The designed aminocoumarin analogues were optimized in Gauss View with DFT, employing Becke's exchange functional combining Lee, Yang, and Parr's (LYP) correlation functional [11, 12]. The STO-3G basis set was used. The STO-3G basis set is a minimal (1s,2s,2P) basis and can be applied to large organic molecules [13].
Protein Selection
The Protein Data Bank (PDB) is a worldwide source of experimentally determined 3D structures of proteins, and their complexes with ligands. Experimentally determined 3D structure of DNA gyrase B 24KDa of Staphylococcus aureus complexes with Novobiocin were retrieved from the RCSB Protein Data Bank in PDB file format. The PDB ID of the respective protein is 4URO. DNA gyrase B 24kDa of Staphylococcus aureus consists of four chains: A, B, C, and D. The A chain was chosen for research.
Protein Preparation
Swiss-PDB Viewer was used to prepare the three-dimensional structure of the ATP binding domain of DNA gyrase B of Staphylococcus aureus (PDB ID:4URO). The protein preparation workflow includes adding missing hydrogen atoms, removing co-crystallized water molecules (non-structural), determining the most likely protonation state, optimizing the protein's hydrogen bond network, etc. The computations were done through vacue with the GROMOS96 43B1 parameter set without a reaction field. GROMOS is an acronym for the GROningen Molecular Simulation computer program package, used for the dynamic modeling of macromolecules.
Molecular docking
Molecular docking was performed to predict non-covalent bond interaction between macromolecules (protein) and a small molecule (ligand). Predicting the non-covalent bond interaction of a small molecule to a macromolecule is important for finding drug-like candidates.[14] We used AutoDock Tools (ADT) of the MGL software package of AutodockVina for the conversion of the PDB file format of the ligands into the PDBQT file format as well as proteins. All docking methods use a scoring function for ranking the various candidate binding modes. The AutoDock scoring function was based on AMBER (i.e., a molecular mechanic force field), and the scoring functions were calibrated using Gasteiger charges. The binding affinity of the ligands to the protein was calculated in kcal/mole [15].
Molecular dynamics simulation
Molecular dynamics was run for Validation of docking studies. For molecular dynamics simulation purposes, we used NAMD. The structure produced static information by using NAMD was viewed with VMD (i.e., a molecular graphics software).[16] NAMD works with AMBER and CHARMM force fields. CHARMM General Force Field is an organic force field that simulates drug-like molecules in a biological environment [17]. The Transferable Intermolecular Potential 3 Points (TIP3P) water model was used by adding Cl- and Na+, where the total solvent molecules, 10480 atoms, had a 1.012 gm/cm3 density. A periodic boundary condition was employed to perform the simulation, where the used box size was 46.0×48.6×53.6. The molecular dynamics trajectories were saved 100 picoseconds for analysis.
Pharmacokinetic parameters study
Undesirable pharmacokinetics and toxicity of the compounds are the main culprits for the failure of drug candidates at the clinical trials stage. Hence, a compound's absorption, distribution, metabolism, excretion, and toxicity (ADMET) should be calculated as early as possible to find the compound with expected pharmacokinetic properties [18]. There are many servers to predict ADMET, such as Swiss ADME, admet SAR, ADMET lab, etc. The Swiss ADME is a charge-free web tool for predicting drug-likeness, pharmacokinetics, and medicinal chemistry friendliness [19]. The ADMET lab web platform is designed based on the Django framework in Python ADMET evaluation of chemicals based on a comprehensively collected ADMET database of 288,967 entries [20]. We used Swiss ADME and ADMET lab 2.0 servers for pharmacokinetics parameters prediction. At first, we generated the SMILES of the designed analogues by the” Online SMILES Translator and Structure File Generator NCI/CADD,” which is developed by the Nationals Institute of Health (NIH), then submitted the generated smiles into the Swiss ADME, and ADMET lab 2.0 servers for pharmacokinetic parameters prediction.
Results and Discussions
Strategies of the designed analogues
Figure 1: Aminocoumarin skeleton
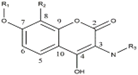
The Figure 1. is the aminocoumarin derivatives scaffold where R1, R2, and R3 are the main modifiable positions. Moreover, the 5th and 6th positions are metabolic sites that could be blocked for reducing toxicity.
Table 1: The added moieties at the modified positions and metabolic sites for designing aminocoumarin analogues.
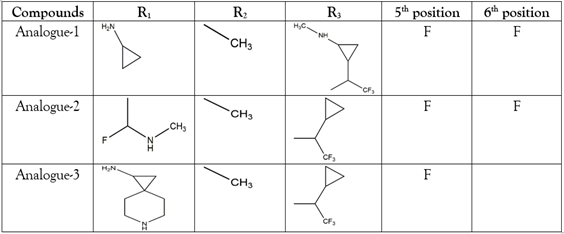
The Amine group at position 3 of the aminocoumarin skeleton forms amide bond in the Novobiocin, Coumermycin, and Clorobiocin. Predicting the good solubility and oral-bioavailability profile of designed analogues requires the modification of the basic fragments. For this reason, we changed the amide bond by its bioisosteres trifluoroethylamine in the designed three analogues, which were shown in Table 1. The electron-withdrawing effect of the CF3 in the trifluoroethylamine declines the basicity of the amine. Moreover, it produces non-basic amine, enabling a good noncovalent bond with the targeted enzyme [21]. The metabolic instability of any compound is an issue. Methyl and methoxy groups in the benzene ring are identified as the metabolically liable sites. The oxidation reaction occurs at metabolic sites by the liver enzymes cytochrome P450, etc. Metabolically liable sites could be blocked by substituting them with Fluorine. For instance, the methyl group is substituted by CF to inhibit the oxidation [22]. We added Fluorine at the 5th and 6th positions to the aminocoumarin skeleton as a metabolic site blocker for designing analogues shown in Table 1. Moreover, we added alicyclic, aliphatic moieties that contained sp3 hybridized carbon atoms at R1, R2, and R3 positions of the aminocoumarin skeleton for designing analogues shown in Table 1. The sp3 hybridized containing organic fragments are saturated. The saturation links with the solubility of a compound. The higher the degree of saturation of a compound, the higher the degree of solubility. The solubility of a compound is measured by the Fsp3 formula, Where Fsp3 is the number of sp3 hybridized carbon/total carbon atoms [23].
Optimization of designed analogues
Since the conformational features of molecules significantly impact their physical and chemical properties, all of the designed analogues were subjected to complete geometry optimization using DFT.
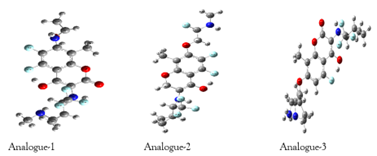
Figure 2: The optimized structures of designed analogues.
The designed analogues of aminocoumarin were optimized in the gas phase at B3LYP/STO-3G level in Gaussian 09. The optimized structure of designed aminocoumarin analogues is shown in Figure 2.
Table 2: The electronic energy (in Hartree) and dipole moment (Debye) of the designed analogues calculated through Gaussian 09W.
| Compounds | Electronic Energy (Hartree) | Dipole Moment (Debye) |
| Analogue-1 | -1677.753506 | 2.7754837 |
| Analogue-2 | -1610.338155 | 1.375769 |
| Analogue-3 | -1695.342940 | 4.413761 |
According to Table 2. data, the highest electronic energy obtained Analogue -2, and the most elevated dipole moment (i.e., represent polarity in nature) was shown in Analogue-3. The highest dipole moment value of Analogue-3 predicts that it will be more soluble.
Molecular docking analysis
Table 3: Binding affinity (kcal/mol) and nonbonding interaction of Novobiocin and designed analogues.
| Compounds | Binding affinity (kcal/mol) | Hydrophobic bonds | Electrostatic bonds | Hydrogen and Halogen bonds | ||||||
| Bonding types | Interacting Amino acids | Distance (Å) | Bonding types | Interacting Amino acids | Distance (Å) | Bonding types | Interacting Amino acids | Distance (Å) | ||
| Novobiocin | -7.2 | Pi-cation | Arg84 | 4.42758 | Conventional Hydrogen bond | Asn54 | 2.19733 | |||
| Analogue-1 | -7.1 | Amide Pi-stacked | Asn54 | 5.50015 | Conventional Hydrogen bond | Arg84 | 2.91847 | |||
| Halogen bond | Asp81 | 3.01531 | ||||||||
| Analogue-2 | -7.4 | Conventional Hydrogen bond | Arg84 | 2.96409 | ||||||
| Halogen bond | Asn54 | 3.4048 | ||||||||
| Halogen bond | Asp81 | 3.03626 | ||||||||
| Analogue-3 | -7.7 | Pi-Cation | Arg84 | 3.98805 | Conventional Hydrogen bond | Asn54 | 2.21785 | |||
| Halogen bond | Asp81 | 3.14878 | ||||||||
We performed side-specific rigid docking of all designed analogues into the binding site amino acids Asn54, Asp81, and Arg84 of DNA gyrase of Staphylococcus aureus using Auto dock vina at identical optimized docking conditions. The binding affinity of Novobiocin and designed aminocoumarin analogues ranged from -7.1 kcal/mol to -7.7 kcal/mol shown in Table 3. According to Table 3 data, Analogue-1 had a lower binding affinity than novobiocin, and Analogue-2 and Analogue-3 showed the highest binding affinity compared to novobiocin. Novobiocin showed pi-cation interaction with Arg84 and conventional hydrogen bond interaction with Asn54. It was seen that Novobiocin skipped to bind one active site amino acid: Asp81. The analogue-1 showed hydrophobic amide-pi-stacked interaction with the Asn54. It also formed conventional hydrogen bond interaction with Arg84 and halogen bond interaction with Asp81. Analogue-2 formed a conventional hydrogen bond with Arg84 and halogen bond interactions with Asn54 and Asp81. Analogue-3 formed electrostatic pi-cation interaction with Arg84 and conventional hydrogen and Halogen bond interactions with Asn54 and Asp81, respectively.
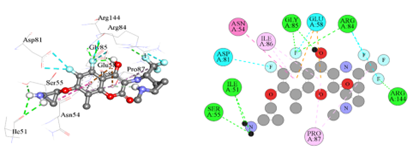
Figure 3(a): The binding orientation map of important amino acids for analogue-1.
The Figure 3(a) showed that Analogue-1 formed Hydrogen bond interaction (green), Halogen bond interaction (Cyan), amide-pi-stacked interaction (pink).
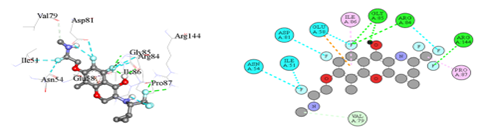
Figure 3(b): The binding orientation map of important amino acids for analogue-2.
The Figure 3(b) showed that Analogue-2 formed Hydrogen bond interaction (green color), Halogen bond interaction (Cyan).
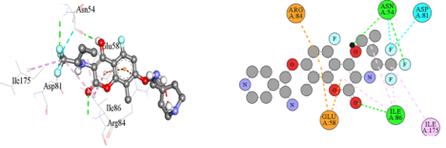
Figure 3(c): The binding orientation map of important amino acids for analogue-3.
The Figure 3(c) showed that Analogue-3 formed Hydrogen bond interaction (green color), Halogen bond interaction (Cyan), and Pi-cation interaction (orange).
Molecular Dynamics simulation
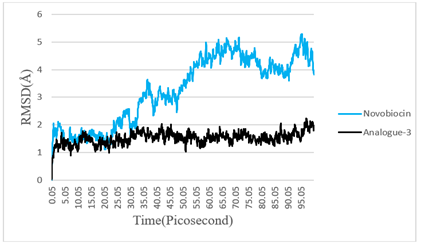
Figure 4: The time series of the RMSD of Novobiocin and Analogue-3 during Molecular Dynamics simulation.
To understand the binding mechanism, structural behavior, and flexibility of Analogue-3, we performed molecular dynamics (MD) simulations for 100 picoseconds. Novobiocin was also subjected to MD simulation as a reference compound. The atomic RMSD for both Novobiocin and the analpgue-3 were calculated and plotted in a time-dependent manner shown in Figure 4. It was seen in Figure 4 that Novobiocin was stable till 30 picoseconds then Novobiocin showed high fluctuation. But analogue-3 showed its stability up to 90 picoseconds.
Drug likeness analysis
Table 4: Drug-likeness analysis of designed analogues and Novobiocin.
| Compounds | Lipskinˈs rule of five | Used server | |||
| Molecular mass < or> | (C Log P) < or> | Number of hydrogen bond donors < or> | Number of hydrogen bond acceptors < or> | ||
| Novobiocin | 612.62 | 2.69 | 5 | 11 | SwissADME |
| Analogue-1 | 439.37 | 2.01 | 4 | 11 | SwissADME |
| Analogue-2 | 426.35 | 2.37 | 3 | 10 | SwissADME |
| Analogue-3 | 471.45 | 2.27 | 4 | 10 | SwissADME |
Oral drug-like compounds follow Lipskin's rule of five: molecular weight not more than 500 g/mol, hydrogen bond acceptor (H-bond acceptor) not more than 10, hydrogen bond donors (H-bond donors) not more than 5, and calculated LogP not more than 5. [24] Any drug that meets Lipskin's rule of five is considered to have good oral bioavailability. [25] According to the Table 4 data, the predicted values of H-bond donors, H-bond acceptor, Molecular weight, and ClogP of Analogue-2 and Analogue-3 followed Lipskin's rule of five.
Selected pharmacokinetics parameter study
Table 5: Pharmacokinetic parameters of novobiocin and designed analogues.
| Parameters | Novobiocin | Analogue-1 | Analogue-2 | Analogue-3 |
| Blood-brain barrier penetration | No | No | No | No |
| GI absorption | low | High | High | High |
| CYP12A inhibitor | No | No | No | No |
| CYP2C19 inhibitor | No | No | No | No |
| CYP2C9 inhibitor | No | No | No | No |
Several pharmacokinetics parameters such as gastrointestinal absorption (GI absorption), Blood-brain barrier (BBB) penetration, CYP1A2 inhibitor, CYP2C19 inhibitor, and CYP2C9 Inhibitor, which values were predicted for designed analogues as well as Novobiocin via SwissADME shown in Table 5. According to Table 5 data, the designed analogues will be showed less toxicity high GI absorption.
Solubility analysis
Carbon bond saturation is represented by Fraction sp3 (Fsp3). The Fsp3 formula is- Fsp3=number of sp3 hybridized carbon ÷ total carbon count. The saturation of carbon bonds is connected with solubility [26].
Table 6: Fsp3 value of Novobiocin and designed analogues
| Compounds | Fsp3 value | Used server |
| Novobiocin | 0.387 | ADMETlab 2.0 |
| Analogue-1 | 0.526 | ADMETlab 2.0 |
| Analogue-2 | 0.556 | ADMETlab 2.0 |
| Analogue-3 | 0.591 | ADMETlab 2.0 |
The predicted Fsp3 values of the design analogues shown in Table 6 indicated that the designed analogues will be more saturated and more soluble than Novobiocin.
Discussion
Molecular interaction analysis of the docking simulation exposed that analogue-3 will be a potent inhibitor of DNA gyrase B of Staphylococcus aureus because it effectively blocked the active site amino acids by non-bond interaction such as conventional hydrogen bond interaction with the Asn54, halogen bond interaction with the Asp81, and pi-cation interaction with the Arg84. After MD data analysis, it was seen that Analogue-3 had the more stability than Novobiocin. Drug likeness analysis suggested that the analogue-3 followed all the parameter of Lipskin’s rule of five. Moreover, the design analogues contained a good predicted value of pharmacokinetic parameters. Finally, it was predicted that the designing of aminocoumarin analogues by converting amide bond of the aminocoumarin skeleton into its bioisosteres trifluoroethylamine, blocked the metabolic sites: the 5th and 6th positions of the aminocoumarin skeleton by fluorine, and tagged saturated aliphatic, alicyclic organic fragments to the aminocoumarin skeleton, will be an excellent approach to solve aminocoumarin derivatives issues.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Md. Al-Amin, and Md. Anawarul Haque permitted unrestricted use, distribution, and reproduction of the information of this article in any medium, provided the original author and source are credited.
Availability of data and materials
All data supporting the findings of this research are available within the article and its supplementary materials files.
Competing interests
Md. Al-Amin, and Md. Anawarul Haque have declared that no competing interests exist.
Author Contributions
Conceptualization: Md. Al-Amin. Data curation: Md. Al-Amin. Formal analysis: Md. Al-Amin. Investigation: Md. Al-Amin. Methodology: Md. Al-Amin. Project administration: Md. Anawarul Haque. Supervision: Md. Anawarul Haque. Validation: Md. Al-Amin. Visualization: Md. Al-Amin. Writing – original draft: Md. Al-Amin. Writing – review & editing: Md. Anawarul Haque.
Funding
Md. Al-Amin, and Md. Anawarul Haque did not received any funding for this work.
References
- Heide, L. (2009). The aminocoumarin: Biosynthesis and biology. Natural Products Reports, 26:1241-1250.
Publisher | Google Scholor - Willcocks, S. J., Cia, F., Francisco, A. F., & Wren, B.W. (2020). Revisiting aminocoumarins for the treatment of melioidosis. International Journal of Antimicrobial Agents, 56(1).
Publisher | Google Scholor - Martindale, W. (1993). Novobiocin. In J.E. Reynolids (Eds.), Martindale: The Extra Pharmacopoeia (30th ed., pp.189). Pharmaceutical Press.
Publisher | Google Scholor - Anderle, C., Stieger, M., Burrell, M., Reinelt, S., Maxwell, A., et al. (2008). Biological activities of novel gyrase inhibitors of the aminocoumarin class. Antimicrobial agents and chemotherapy, 52(6):1982–1990.
Publisher | Google Scholor - Maxwell, A. (1997) DNA gyrase as a drug target. Trends in microbiology, 5(3):102-108.
Publisher | Google Scholor - Maxwell, A., & Lawson, D. M. (2003). The ATP-binding site of type ii topoisomerases as a target for antibacterial drugs. Current Topics in Medicinal Chemistry, 3(3):283-303.
Publisher | Google Scholor - Gilbert, J. E., & Maxwell, A. (1994). The 24 kDa N-terminal sub-domain of the DNA gyrase B protein binds coumarin drugs. Molecular Microbiology, 12(3):365-373.
Publisher | Google Scholor - Kowalczyk, A., Paneth, A., Trojanowski, D., Paneth, P., Zakrzewska-Czerwinska, J., et al. (2021). Thiosemicarbazide derivatives decrease the ATPase activity of staphylococcus aureus topoisomerase iv, inhibit mycobacterial growth, and affect replication in mycobacterium smegmatis. International Journal of Molecular Science, 22:3881.
Publisher | Google Scholor - Laurin, P., Ferroud, D., Klich, M., Dupuis-Hamelin, C., Mauvais, P., et al. (1999). Synthesis and in vitro evaluation of novel highly potent coumarin inhibitors of gyrase B. Bioorganic & Medicinal Chemistry Letters, 9(14):2079-2084.
Publisher | Google Scholor - Tong, S., Davis, J., Eichenberger, E., Holland, T., & Flower Jr, V. (2015). Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clinical Microbiology Reviews, 28(3):604.
Publisher | Google Scholor - Lee, C., Yang, W., Parr, RG. (1998). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Physical Review B,37(2):785.
Publisher | Google Scholor - Becke, AD. (1988). Density-functional exchange-energy approximation with correct asymptotic behavior. Physical Review A, 38(6):3098.
Publisher | Google Scholor - Hehre, W. J., Stewart, R. F., & Pople, J. A. (1969). Self-Consistent molecular-orbital methods. I.Use of Gaussian expansions of Slater-Type atomic orbitals. The Journal of Chemical Physics, 51(6):2657-2664.
Publisher | Google Scholor - Trott, O., & Olson A. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2):455–461.
Publisher | Google Scholor - Morris, G., & Lim-Wilby, M. (2008). Molecular docking. In. A. Kukol, & Walker, JM (Eds.), Molecular Modelling of Proteins (2nd ed. pp. 365-382). Humana Press.
Publisher | Google Scholor - Phillips, J. C., Braun, R., Wang, W., Gumbart, J., Tajkhorshid, E., et al. (2005). Scalable molecular dynamics with NAMD. Journal of Computational Chemistry, 26(16):1781-1802.
Publisher | Google Scholor - Vanommeslaeghe, k., Hatcher, E., Acharya, C., Kundu, S., Zhong, S., et al. (2009). CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. Journal of Computational Chemistry, 31:671-690.
Publisher | Google Scholor - Jia, C., Li, J., Hao, G., & Yang G. (2020). A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discovery Today, 25(1):248-258.
Publisher | Google Scholor - Daina, A., Michielin, O., & Zoete, V. (2017). SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports.
Publisher | Google Scholor - Dong, J., Wang, N., Yao, Z., Zhang, L., Cheng, Y., et al. (2018). ADMETlab: A Platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. Journal of Cheminformatics, 10(29).
Publisher | Google Scholor - Black, W., Bayly, C., Davis, D., Desmarais, S., Falgueyret, J., et al. (2005). Trifouroethylamines as amide isosteres in inhibitor of cathepsin k. Bioorganic & Medicinal Chemistry Letters, 15(21):4741-4744.
Publisher | Google Scholor - Bohm, H., Banner, D., Bendels, S., Kansy, M., Kuhn, B., Muller, k., Obst-Sander, U., & Stahl, M. (2004). Fluorine in medicinal chemistry. Chem Bio Chem, 5:637-643.
Publisher | Google Scholor - Aldeghi, M., Malhotra, S., Selwood, D., & Chan, A. (2013). Two- and three-dimensional rings in drugs. Chemical Biology & Drug Design, 83:450-461.
Publisher | Google Scholor - Lipinski, C. A., Lombardo, F., Dominy, B. W., & Feeney, P.J. (1997). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews, 23:3-25.
Publisher | Google Scholor - Tijjani, H., Olatunde, A., Adegunloye, P.A., & Ishola, A. A. (2022). In silico insight into the interaction of 4-aminoquinolines with selected SARS-CoV-2 structural and nonstructural proteins. Coronavirus Drug Discovery, 3:313-333.
Publisher | Google Scholor - Lovering, F., Bikker, J., & Humblet, C. (2009). Escape from flatland: Increasing saturation as an approach to improving clinical success. Journal of Medicinal Chemistry, 52(21):6752-6756.
Publisher | Google Scholor












