

Research Article
Comprehensive Design and Evaluation of Fast-Disintegrating Tablets Using Box-Behnken Statistical Approach
- Tushar Sonare
- Aman Kumar
- Kratika Khadsondni
- Krutika Mandloi
- Akash Yadav *
- Dinesh Kumar Jain
IPS Academy College of Pharmacy, Knowledge Village, Rajendra Nagar, A.B. Road, Indore-452012, India.
*Corresponding Author: Akash Yadav, IPS Academy College of Pharmacy, Knowledge Village, Rajendra Nagar, A.B. Road, Indore-452012, India.
Citation: Sonare T., Kumar A., Khadsondni K., Mandloi K., Yadav A., et al. (2025). Comprehensive Design and Evaluation of Fast-Disintegrating Tablets Using Box-Behnken Statistical Approach, Journal of Clinical Research and Clinical Trials, BioRes Scientia Publishers. 4(3):1-15. DOI: 10.59657/2837-7184.brs.25.048
Copyright: © 2025 Akash Yadav, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: February 07, 2025 | Accepted: February 25, 2025 | Published: March 03, 2025
Abstract
This study thoroughly examined the development of rapidly disintegrating glipizide tablets, employing a Box–Behnken design along with the direct compression technique. The research specifically focused on the use of super-disintegrants namely cross-povidone and sodium starch glycolate in combination with the binder hydroxypropyl methylcellulose. Extensive pre-formulation assessments were conducted to verify the physicochemical compatibility between glipizide and the selected excipients. The produced fast-dissolving tablets underwent a detailed series of evaluations, including tests for weight variation, mechanical strength, friability, disintegration time, in vitro drug release behaviour, and stability. The strategic use of the Box–Behnken design was effective in optimizing the proportions of the synthetic super-disintegrants, ensuring quicker disintegration and an ideal release profile for the active pharmaceutical ingredient. This innovative formulation of fast-dissolving glipizide tablets is expected to significantly improve patient adherence, especially among diabetic patients experiencing swallowing difficulties, potentially enhancing treatment effectiveness and overall disease control.
Keywords: glipizide; box-behnken design; rapidly disintegrating
Introduction
Diabetes mellitus a chronic metabolic condition marked by elevated blood sugar levels, which can harm organs such as the liver, kidneys, eyes, and blood vessels over time. There are two primary forms: Type 1 and Type 2. Type 2 diabetes is the more prevalent type, mainly caused by insulin resistance or insufficient insulin production. It is being increasingly diagnosed among younger individuals due to rising rates of obesity, unhealthy diets, and a lack of physical exercise. In recent decades, the incidence of Type 2 diabetes has surged worldwide across all economic levels. Type 1 diabetes, an autoimmune disorder, generally arises during childhood or adolescence and leads to minimal or no insulin production. Managing both types of diabetes necessitate ongoing access to healthcare and insulin to prevent complications. With approximately 1.5 million fatalities each year, diabetes represents a major global health challenge. Addressing these epidemic calls for concerted global efforts to enhance healthcare, decrease obesity, and encourage healthier lifestyle choices by 2025 [1-2].
Glipizide is a type of sulfonylurea that is utilized in the treatment of 2 diabetes mellitus by enhancing insulin production and increasing sensitivity to insulin. With a molecular weight of 445.536 g/mol, it reduces blood glucose levels by prompting the pancreas to generate more insulin. Glipizide comes in various forms, including sustained-release tablets and oral solutions, and is included in important pharmacopoeias such as the Indian Pharmacopoeia (IP), British Pharmacopoeia (BP), and United States Pharmacopoeia (USP). This medication is typically administered orally, and it has a biological half-life of 2 to 6 hours. Glipizide is mainly used for managing non-insulin-dependent diabetes (Type 2), providing a reliable way to control elevated blood sugar levels and enhancing the effectiveness of insulin, making it a crucial component in the management of Type 2 diabetes [3-4].
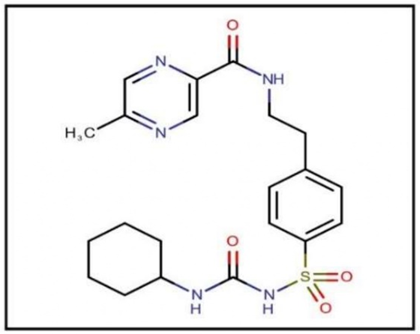
Figure 1: Glipizide.
Fast Dissolving Tablets
Fast-dissolving tablets (FDTs) are innovative oral dosage forms designed to break down and release their active ingredients almost immediately when they come into contact with the oral mucosa, eliminating the need for water. These tablets are crafted to dissolve in just a few seconds to a minute, greatly enhancing patient compliance, especially for those with swallowing difficulties or dysphagia. FDTs often utilize super disintegrants and are commonly used in situations that require a quick therapeutic effect, such as treating acute pain, nausea, or allergic reactions.
Ideal Characteristics of Fast Dissolving Tablets
- The tablet should break down quickly, within seconds to a minute, when placed in the mouth, without n q qeeding water for consumption.
- It is specifically designed for people with swallowing difficulties, promoting better adherence to therapy and allowing for easy administration.
- The formulation must allow for the rapid release of the active pharmaceutical ingredient (API), ensuring a fast onset of therapeutic action.
- The tablet should be appropriately sized for easy handling and swallowing, prioritizing convenience for users, especially those with special needs.
- The formulation needs to be carefully crafted to enhance the availability of the active ingredient, allowing for prompt therapeutic response.
Advantages
- Conventional dose forms require liquid for ingestion, whereas FDTs do not.
- This makes it easier for patients who travel or don't have water to comply.
- The bioavailability of insoluble medications is improved by rapid pill dissolving and disintegration.
- Conventional processing and packaging equipment allow the manufacturing of tablets at low cost.
Tablets dissolve and absorb quickly in the mouth [5-6].

Figure 2: Fast dissolving tablet.
Box-Behnken Experimental Design
Response Surface Methodology (RSM) is centered on examining the connection between control variables and response variables to enhance processes. An essential element of RSM is the careful design of experiments, which aids in reducing costs while considering factors like mixtures and levels. Several experimental designs, such as full factorial, Box-Behnken Design (BBD), and central composite designs, play a crucial role in optimization research. The Box-Behnken Design, frequently utilized in RSM and accessible through software like Design Expert (version 7.0, Stat-Ease Inc.), is particularly useful for investigating three-level experiments. It generally includes 15 experimental trials, alongside five repeated runs to guarantee trustworthy results and consistency. This design employs a second-order polynomial model to depict the relationships between input and output variables. By leveraging these experimental designs, researchers can expedite the optimization of processes in areas like pharmaceuticals. These designs enhance data collection and analysis, making the process more efficient and ultimately boosting research productivity. The Box-Behnken Design, in particular, is appreciated for its capacity to deliver dependable insights using fewer trials, thereby conserving resources while obtaining valuable information. In summary, utilizing RSM and effective experimental designs improves the speed and efficacy of research, aiding in the development of optimized processes and solutions [7-9].
Selected Independent Variables and Dependent Variables
The selected independent variables are displayed in Table 1.
Table 1: Independent variables with levels.
| Factor | Name | Unit | Minimum | Maximum |
| A | Cross-Povidone | % | 1 | 4 |
| B | Sodium Starch Glycolate | % | 2 | 10 |
| C | Hydroxy Propyl Methylcellulose | % | 1 | 5 |
The selected Dependent variables are displayed in Table 2.
Table 2: Dependent Variables.
| Response | Variables | Unit |
| 1. | Dissolution | (%) |
| 2. | Disintegration time | Seconds |
| 3. | Hardness | Kg/cm2 |
Table 3: Formulation Runs as per Box-Behnken Design.
| Std | Runs | Independent Variables | Dependent Variables | ||||
| Cross-Povidone (mg) | Sodium Starch Glycolate (mg) | Hydroxy Propyl Methylcellulose (mg) | Dissolution (%) | Disintegration (Second) | Hardness (Kg/cm2) | ||
| 13 | 1 | 2.5 | 6 | 3 | 93.81 | 19.43 | 2.6 |
| 12 | 2 | 2.5 | 2 | 3 | 94.82 | 21.86 | 2.9 |
| 5 | 3 | 4 | 6 | 1 | 90.17 | 16.78 | 2.1 |
| 2 | 4 | 2.5 | 2 | 5 | 96.34 | 22.03 | 2.4 |
| 4 | 5 | 4 | 10 | 3 | 98.43 | 14.23 | 2.3 |
| 6 | 6 | 2.5 | 2 | 1 | 95.17 | 21.29 | 2.1 |
| 10 | 7 | 1 | 6 | 1 | 95.43 | 22.65 | 2.2 |
| 1 | 8 | 1 | 10 | 3 | 91.39 | 18.31 | 2.4 |
| 15 | 9 | 2.5 | 10 | 5 | 94.82 | 16.27 | 2.3 |
| 14 | 10 | 1 | 6 | 5 | 93.12 | 21.89 | 2.6 |
| 8 | 11 | 1 | 2 | 3 | 97.47 | 22.23 | 2.8 |
| 3 | 12 | 4 | 6 | 5 | 95.17 | 16.42 | 2.4 |
| 7 | 13 | 2.5 | 6 | 3 | 93.27 | 19.12 | 2.8 |
| 11 | 14 | 2.5 | 10 | 1 | 91.65 | 16.10 | 2.9 |
| 9 | 15 | 2.5 | 6 | 3 | 92.87 | 19.82 | 2.3 |
Materials and Methods
Glipizide was provided as a sample by USV Private Limited, located in the IInd Stage of Peenya Industrial Estate, Bangalore, Karnataka. Microcrystalline cellulose was sourced from Maple Biotech Pvt. Ltd., Pune, Maharashtra, while saccharin was obtained from Ranbaxy, New Delhi. Talc and magnesium stearate were supplied by S.D. Fine Chem Ltd., Mumbai, Maharashtra. All other solvents and reagents for the formulation were procured from the market. These ingredients were carefully selected from reputable suppliers to ensure the final product's quality and effectiveness, maintaining high standards within the pharmaceutical industry. The attention to sourcing and quality control emphasizes the commitment to producing reliable and effective medications for end-users.
Table 4: Physical and Chemical Parameters of Glipizide.
| S. No. | Parameter | Predicted value |
| 1 | Molecular formula | C21H27N5O4S |
| 2 | Molecular structure | 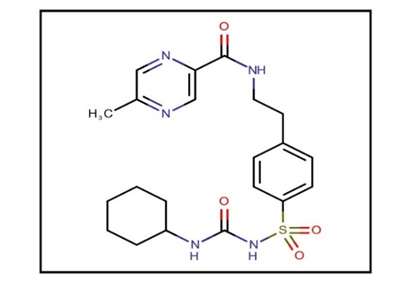 |
| 3 | IUPAC name | N-(4-[N-(cyclohexyl carbamoyl) sulfonyl] phenethyl)-5-methylpyrazine-2-carboxamide |
| 4 | Molecular weight | 445.536 g/mol |
| 5 | BCS class | Class II |
| 6 | pH and pKa | 5.9 and 6.0 |
| 7 | Log P | 1.91 or 1.83 |
| 8 | Crystallinity | Crystalline solid |
| 9 | Melting point | 208-209 0C (406 to 408 °F) |
| 10 | Solubility | Soluble in organic solvents like methanol, acetone |
Pre-Compression Parameters
Angle of Repose: The maximum angle at which a build-up of loose powder remains stable without collapsing is known as the angle of repose. It acts as a gauge of the cohesiveness and flow properties of the powder. Better flow is indicated by lower angles, and worse flow is indicated by greater angles. A mound of powder is progressively built up on a level surface until it starts to slump in order to be measured.

Bulk Density: The mass of a powder divided by its whole volume, which includes the empty spaces between particles, is known as its bulk density. It is commonly given in quantities such as g/cm³ or kg/L and indicates how well the powder packs together. Using tools like a graduated cylinder, a known volume of the powder-including its vacant spaces-must be weighed in order to make a determination.

Tapped Density: The maximum bulk density attained when a powder is tapped or vibrated to remove interparticle gaps is known as "tapped density." The powder is tapped repeatedly in a container until no more volume loss takes place, indicating the powder's packing capabilities under compaction.

Hausner's Ratio: Hausner's ratio is the ratio of tapped density to bulk density, calculated using the formula:

It offers information about the packing efficiency and flow behavior of the powder. Good flowability is indicated by a ratio near 1, and poor flowability is indicated by values greater than 1.
Carr's Index: Carr's index, or compressibility index, quantifies the powder's compressibility and is calculated as follows:

It shows the extent of volume reduction brought about by compression or tapping. Superior flowability is indicated by lower Carr's index values, whereas reduced flow or enhanced cohesion are implied by higher values.
Partition Coefficient: Glipizide, a second-generation sulfonylurea utilized for the treatment of type 2 diabetes, is a lipophilic substance with limited solubility in water. Its partition coefficient (Log P) is between 2.4 and 2.6, suggesting that it dissolves better in lipid environments compared to aqueous solutions. characteristic facilitates its passage through lipid membranes, enhancing oral absorption. However, an excessive degree of lipophilicity could impede its dissolution and bioavailability. In laboratory settings, the partition coefficient of Glipizide is measured by equilibrating it in n-Octanol and water, subsequently calculating the ratio of concentrations in both phases.

Glipizide Identification Studies: Glipizide 10mg was accurately weighed and dissolved in 10ml methanol and subsequent dilution was made to get required concentrations (1000µg/ml). The wave length of maximum absorbance (λmax) of this clear solution was determined from 200-400nm methanol used as blank.
Preparation of Stock Solution: Glipizide 10 mg was dissolved in methanol 10 ml (1000 µg/ml) Stock solution I. From this solution diluted with methanol up to 100ml (100 µg/ml) Stock solution II was prepared.
Preparation of Sample Solution: Precise aliquots of 0.5, 1, 1.5, 2, and 2.5 ml were meticulously drawn from Stock Solution II and subsequently diluted to a final volume of 10 ml with a suitable solvent. This process yielded solutions with concentrations of 5, 10, 15, 20, and 25 µg/ml, respectively. The absorbance of each solution was measured at 268 nm using a UV-visible spectrophotometer (Model 1800), with each reading carefully compared against an appropriate blank to ensure accuracy and minimize deviations. The recorded absorbance values were systematically documented for further evaluation. A standard calibration curve was constructed by plotting the known concentrations on the X-axis against their corresponding absorbance values on the Y-axis. This graphical representation delineated a linear correlation between concentration and absorbance, conforming to Beer-Lambert’s principle. The resulting equation of the calibration curve enabled the precise determination of unknown concentrations by interpolating their absorbance values within the established range. This analytical approach guarantees exceptional accuracy and reproducibility in quantitative assessments, making it highly applicable in pharmaceutical and scientific research. The developed calibration model serves as a fundamental reference for subsequent spectrophotometric analyses, thereby reinforcing the reliability and precision of experimental outcomes.
Melting Point of Glipizide: The melting point of Glipizide was established using an electric melting point device. A capillary tube, sealed at one end, was filled with the drug sample and positioned in the appropriate slot of the apparatus. The device was turned on, and the tube was monitored closely through the built-in magnifying glass for any indication of melting. The temperature at which the sample-initiated melting was carefully recorded from the thermometer. This initial temperature was noted as the melting point of the sample. To guarantee accuracy and reliability, the procedure was repeated three times, and the average of the three measurements was taken as the definitive melting point value. This technique yielded dependable and precise results for characterizing Glipizide [10-14].
Table 5: Formulation of Fast Dissolving Tablets.
| Aspect | Fast dissolving tablets |
| Purpose | To Provide Rapid Dissolution and Absorption for Fast Onset of Action. |
| Drug Release Mechanism | Drug Dissolves Quickly in The Mouth or Stomach. |
| Excipients | Super-Disintegrants- Sodium Starch Glycolate, Cross-Povidone. |
| Binders-Guar Gum, PVPK-30. | |
| Lubricants-Magnesium Stearate. | |
| Glidant-Talc. | |
| Tablet size | Smaller and Thinner Due to Rapid Dissolution Mechanism. |
| Tablet Compression | Needs to be Compressed Under Low Force to Ensure Quick Dissolution. |
| Drug Release Profile | Rapid Drug Release Within Minutes, Typically Within 3-5 Minutes of Contact with Water or Saliva. |
| Preparation Methods | Direct Compression or Molding. |
| Advantages | Fast Onset of Action for Immediate Relief. |
| Convenient For Patients with Swallowing Difficulties. | |
| Enhanced Patient Compliance for Medications That Need to be Taken Urgently. |
Table 6: Pharmacokinetic.
| Pharmacokinetic Parameter | Fast Dissolving Tablets |
| Onset of Action | Faster Onset Due to Rapid Dissolution and Absorption |
| Time to Peak Concentration (Tmax) | 30-60 Minutes (Faster Absorption) |
| Bioavailability | 85-95% (Similar to Conventional Tablets, But May Have Slight Improvement in Absorption Due to Faster Dissolution) |
| Peak Plasma Concentration (Cmax) | Achieved Faster |
| Volume of Distribution (Vd) | 8-10 L/kg |
| Plasma Protein Binding | 98-99% |
Table 7: List of Excipients Used in Formulations and Their Role.
| Excipients | Role |
| Glipizide | Active Pharmaceutical Ingredients |
| Cross-Povidone | Super Disintegrants |
| Sodium Starch Glycolate | Super Disintegrants |
| Hydroxypropyl Methylcellulose | Binder |
| Talc | Glidant |
| Magnesium Stearate | Lubricant |
| Sodium saccharine | Sweating Agent |
| Microcrystalline Cellulose | Filler |
Post-compression Studies
Organoleptic Characters: Each batch of tablets is evaluated for sensory properties, including appearance, color, odor and shape, through direct observation and analysis.
Thickness and Diameter: Measure tablet thickness and diameter length using vernier caliper. Five tablets were selected from each batch and their measurements averaged to give a representative value expressed in millimeters.
Hardness: Test the hardness of the tablets using the Monsanto hardness tester. Three pieces were selected from each map and the average hardness value was recorded. These values are expressed in kilograms per square centimeter (kg/cm2).
Friability: Friability of tablets was assessed using Roche friability tester as per the guidelines prescribed in Indian Pharmacopoeia (IP) standards. Initially, the weight of 20 pieces was recorded as the starting weight (Wi). Set the grinder to run at 25 revolutions per minute (rpm) for 4 minutes. Thereafter the tablets were reprocessed (Wf). Then, the friability percentage (%F) of each batch of tablets was calculated using the given formula:

It is worth noting that friability should not exceed 1.0% as per the standards set by the United States Pharmacopoeia (USP), Indian Pharmacopoeia (IP) and British Pharmacopoeia (BP).
Weight Variation: Twenty tablets were selected from each batch before weighing, individual weight was measured and averaged. The weight was measured as per the specifications mentioned in the Indian Pharmacopoeia (I.P.). According to the I.P standard the average difference in weight should not exceed 5% and two tablets and no tablet should exceed two times the percentage.

In-Vitro Disintegration Time: Tablet disintegration time was evaluated using a USP disintegration tester in water maintained at 37 ± 2 °C. As standard partial disintegration, six tablets from each batch were placed in one liter of distilled water. The time required to complete disintegration of all six tablets was recorded. Ideally, the duration of all tablets should not exceed 3 minutes. If more than one tablet failed to meet this criterion, the study was repeated using 12 tablets. It is expected that no more than 2 out of 18 tablets will differ from the unseparated standards.
In-vitro Dissolution Studies: The release rate of Glipizide tablets was evaluated using a USP Type II dissolution apparatus in paddle configuration. Place one tablet in 6 dissolution vials, each containing 900 ml of separation medium, pre-equilibrated at 37 ± 0.5 °C and mixed at 50 rpm. At the appointed time, remove 2 ml aliquots of the dissolution medium and replace them with fresh dissolution medium. The sample was then filtered, diluted, and analyzed spectrophotometrically at 268 nm to determine the concentration of the drug relative to the corresponding concentration. This process is done on all batches. The average percentage of Glipizide released at different time points was determined from the sample image and plotted against time.
Calculation of % drug release involves the following steps:
Determined concentration of drug released by using formula:

Y is absorbance, m is slope, C is intercept, X is concentration (µg/ml).

Stability Studies: In this study, all formulations were evaluated for one-month stability in accordance with the rapid study criteria established by the International Committee for Harmonization (ICH). The sample was carefully wrapped in aluminum foil and stored in airtight glass. The tablets were stored at three different temperatures. Over a period of 10, 20 and 30 days, the tablets are removed from storage and analyzed for physical properties, separation properties and drug content [15-21].
Formulation of Glipizide by Using Direct Compression Method
Direct compression is a method used in tablet manufacturing where the ingredients of the formulation are combined, processed, and then pressed into tablets. This technique is particularly well-suited for tablets that include hygroscopic and thermolabile active pharmaceutical ingredients (APIs). It is preferred because of its straightforwardness and cost-efficiency when compared to other tablet compression methods. In direct compression, excipients and APIs are mixed into a powdered blend, with particle sizes regulated through the use of mills and sieves, and then they are compressed into tablets. This process demands minimal equipment and fewer steps, making it the most efficient and cost-effective approach for tablet production.
Ideal Characteristics of direct compression method
- Reduced processing steps lead to lower production expenses.
- When the formulation is appropriately designed, direct compression can yield tablets that have consistent weight, hardness, and dissolution characteristics.
- This approach needs fewer and simpler machines, making maintenance and operation easier.
- A decrease in handling steps and equipment diminishes the chances of contamination during production.
Various excipients enable flexible formulations to meet diverse product requirements [22-23].
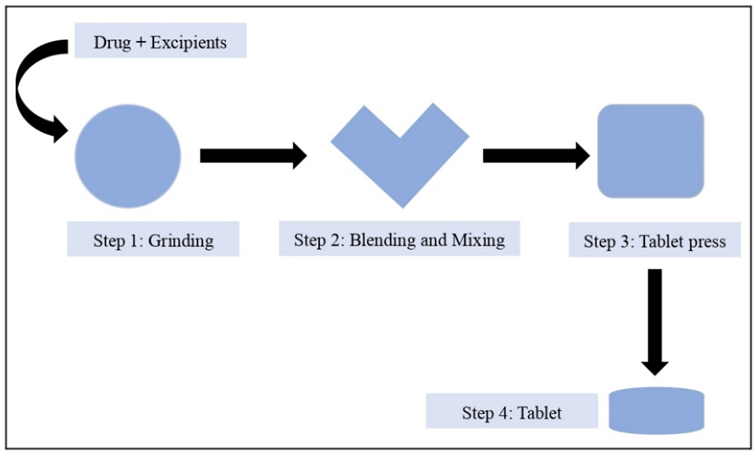
Figure 3: Formulation of Fast Dissolving Tablets by using Direct Compression Method
Result and Discussion
Organoleptic Properties: Organoleptic properties of the drug sample were found to be as given in table below.
Table 8: Organoleptic properties of Glipizide.
| Sl.no | Organoleptic Characteristics | Result |
| 1. | Color | White to Off-White Crystalline Compound |
| 2. | Nature | Crystalline Powder |
| 3. | Odour | Odorless |
| 4. | Taste | Slightly Bitter |
Melting Point of Glipizide: The final reading was obtained by averaging the results of three independent experiments, resulting in a temperature of 209°C.
Partition Coefficient: The partition coefficient of Glipizide is found to be 1.91.
pH determination of Glipizide: The pH range glipizide is adjusted to 6.8 to 7.0 during preparation to ensure that it is stable and effective in treating diabetes.
Solubility Study of Glipizide: A solubility assessment was conducted to examine how Glipizide dissolves in methanol. During this experiment, different quantities of Glipizide were introduced into flasks that contained a consistent volume of methanol. The mixtures were continuously stirred for 24 hours to guarantee that the drug fully dissolved. After the stirring process, the solutions were filtered to eliminate any undissolved Glipizide particles, resulting in a clear solution. The concentration of the dissolved Glipizide was determined using a UV-Vis spectrophotometer, comparing absorbance values with a calibration curve that correlated absorbance to known concentrations. By considering the amount of Glipizide added, the volume of methanol used, and the absorbance data, the solubility was calculated in milligrams per milliliter (mg/mL). This method could also be modified to investigate how temperature and various solvents impact Glipizide's solubility, offering useful insights for formulation development.
Table 9: Solubility of Glipizide in Solvents.
| S. No. | Name of solvent | Solubility |
| 1. | Methanol | Freely Soluble |
| 2. | Ethanol | Practically Soluble |
| 3. | Acetone | Slightly Soluble |
| 4. | Methylene Chloride | Slightly Soluble |
| 5. | Water | Practically Insoluble |
Table 10: Flow Properties of Powder Blend.
| S. No | Angle of Repose (θ) | Bulk Density (g/cm2) | Tapped Density (g/cm2) | Hausner’s Ratio | Carr’s Ratio (%) |
| 1. | 33.26 | 0.52 | 0.64 | 1.23 | 18.40 |
| 2. | 32.55 | 0.47 | 0.53 | 1.12 | 11.35 |
| 3. | 31.74 | 0.52 | 0.60 | 1.15 | 13.38 |
| 4. | 34.45 | 0.48 | 0.57 | 1.16 | 14.03 |
| 5. | 33.23 | 0.55 | 0.64 | 1.12 | 10.93 |
| 6. | 32.66 | 0.52 | 0.62 | 1.16 | 14.51 |
| 7. | 30.36 | 0.48 | 0.55 | 1.14 | 12.77 |
| 8. | 32.54 | 0.51 | 0.59 | 1.15 | 14.85 |
| 9. | 33.18 | 0.49 | 0.62 | 1.15 | 16.92 |
| 10. | 34.47 | 0.48 | 0.57 | 1.15 | 14.02 |
| 11. | 33.23 | 0.53 | 0.65 | 1.23 | 18.40 |
| 12. | 31.74 | 0.52 | 0.59 | 1.16 | 13.32 |
| 13. | 33.22 | 0.56 | 0.64 | 1.11 | 10.92 |
| 14. | 33.25 | 0.52 | 0.65 | 1.22 | 18.48 |
| 15. | 32.52 | 0.51 | 0.58 | 1.19 | 14.84 |
Post-Compression Studies
Organoleptic Characters
Table 11: Organoleptic Properties of Glipizide Tablets.
| S. No. | Organoleptic Characteristics | Result |
| 1. | Color | White to Off-White |
| 2. | Shape | Circular |
| 3. | Odour | Odorless |
| 4. | Taste | Palatably Sweet |
Identification of Glipizide by UV Spectrophotometry
Derivation of Drug Spectrum: The methanol stock solution of Glipizide, with a concentration of 10µg/ml, exhibited a peak absorption wavelength (λmax) at 268 nm. The absorbance observed at this wavelength was measured to be 0.1404.
Preparation of Calibration Curve of Glipizide in Water: The calibration curve of glipizide dissolved in methanol was plotted using a UV spectrophotometer and measured at a wavelength of 268 nm. This curve was created from 5 dilutions of stock solution, each at 5 µg/ml. The resulting curve is shown in Table 13.
Table 12: Calibration Curve Data of Glipizide in Methanol.
| S. No. | Concentration (μg/ml) | Absorbance at 268 |
| 1 | 5 | 0.1404 |
| 2 | 10 | 0.2824 |
| 3 | 15 | 0.4513 |
| 4 | 20 | 0.5914 |
| 5 | 25 | 0.7170 |
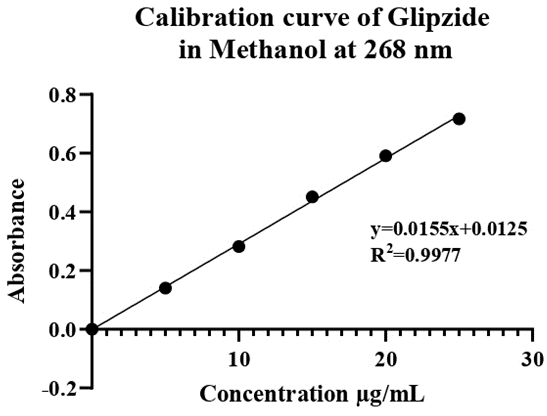
Figure 4: Calibration curve of Glipizide.
Table 13: Post-Compression Parameters of Tablets.
| Formulation Code | Thickness (mm) | Hardness (Kg/cm2) | Friability (%) | Disintegration Time (Sec) |
| FDT 1 | 1.96±0.2 | 1.9 | 0.40 | 20.16 |
| FDT 2 | 2.02±0.2 | 1.8 | 0.42 | 21.48 |
| FDT 3 | 1.99±0.2 | 2.1 | 0.48 | 19.46 |
| FDT 4 | 2.01±0.2 | 1.9 | 0.43 | 20.12 |
| FDT 5 | 2.0±0.2 | 2.3 | 0.44 | 22.12 |
| FDT 6 | 1.98±0.2 | 1.8 | 0.47 | 20.12 |
| FDT 7 | 1.99±0.2 | 1.9 | 0.56 | 19.48 |
| FDT 8 | 2.02±0.2 | 2.0 | 0.58 | 20.12 |
| FDT 9 | 1.98±0.2 | 1.9 | 0.44 | 20.11 |
| FDT 10 | 1.99±0.2 | 1.8 | 0.43 | 18.42 |
| FDT 11 | 2.01±0.2 | 1.9 | 0.47 | 19.49 |
| FDT 12 | 2.00±0.2 | 2.1 | 0.45 | 19.40 |
| FDT 13 | 1.97±0.2 | 2.0 | 0.49 | 20.13 |
| FDT 14 | 1.99±0.2 | 1.8 | 0.48 | 22.16 |
| FDT 15 | 2.01±0.2 | 1.9 | 0.49 | 24.11 |
In-vitro Dissolution Studies
Table 14: Cumulative Drug Release of Glipizide Tablets Formulations (F1-F15).
| Formulation Code | Time (Minutes) | ||||
| 2.5 | 5 | 10 | 15 | 20 | |
| FDT 1 | 40.15 | 58.36 | 82.89 | 92.15 | 96.36 |
| FDT 2 | 38.14 | 59.17 | 81.48 | 90.98 | 95.45 |
| FDT 3 | 93.95 | 57.41 | 81.78 | 91.42 | 95.69 |
| FDT 4 | 43.58 | 58.45 | 83.45 | 93.25 | 97.45 |
| FDT 5 | 41.26 | 57.41 | 81.45 | 90.87 | 94.45 |
| FDT 6 | 42.45 | 58.45 | 83.45 | 93.45 | 94.24 |
| FDT 7 | 46.15 | 60.32 | 88.18 | 98.14 | 99.13 |
| FDT 8 | 51.35 | 62.45 | 89.45 | 99.48 | 99.71 |
| FDT 9 | 49.65 | 59.65 | 85.48 | 95.89 | 98.45 |
| FDT 10 | 50.45 | 58.45 | 84.65 | 97.89 | 98.45 |
| FDT 11 | 40.96 | 57.24 | 81.57 | 92.75 | 96.78 |
| FDT 12 | 42.98 | 58.45 | 83.48 | 93.25 | 94.48 |
| FDT 13 | 42.48 | 58.48 | 84.16 | 93.24 | 94.89 |
| FDT 14 | 46.48 | 59.14 | 88.14 | 98.89 | 98.59 |
| FDT 15 | 51.48 | 62.56 | 89.58 | 99.58 | 99.58 |
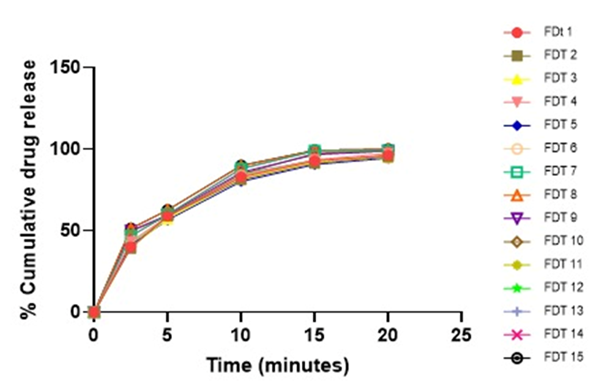
Figure 5: Cumulative drug release of Glipizide Tablets (F1-F15).
Response data for all 15 batches by using Box-Behnken Experimental Design (FI–F15)
Response 1: Dissolution (%)
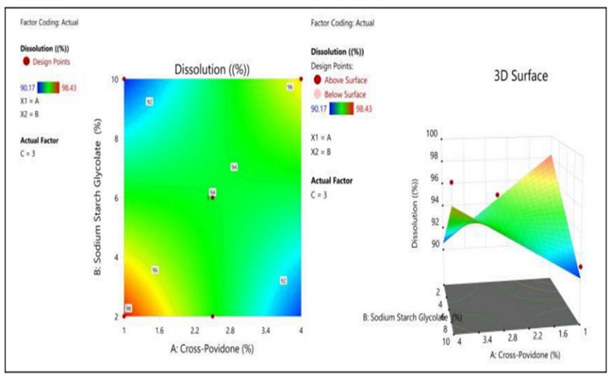
Figure 6: The 2D contour plot and the three-dimensional 3D map shows how the total amounts of super disintegrates (X1) and (X2) affects the dissolution.
ANOVA for Linear model
Response 1: Dissolution.
| Source | Sum of Squares | df | Mean Square | F-value | p-Value | |
| Model | 59.35 | 6 | 9.89 | 6.32 | 0.0102 | Significant |
| A-Cross Povidone | 0.9059 | 1 | 0.9059 | 0.5792 | 0.4684 | |
| B-Sodium Starch Glycolate | 1.54 | 1 | 1.54 | 0.9844 | 0.3502 | |
| C-Hydroxy propyl Methylcellulose | 6.18 | 1 | 6.18 | 3.95 | 0.0821 | |
| AB | 31.41 | 1 | 31.41 | 20.08 | 0.0021 | |
| AC | 13.36 | 1 | 13.36 | 8.54 | 0.0192 | |
| BC | 1.00 | 1 | 1.00 | 0.6394 | 0.4470 | |
| Residual | 12.51 | 8 | 1.56 | |||
| Lack of fit | 12.07 | 6 | 2.01 | 9.04 | 0.1030 | Not Significant |
| Pure Error | 0.4451 | 2 | 0.2225 | |||
| Cor Total | 71.86 | 14 |
Response 2: Disintegrating Time (Second).
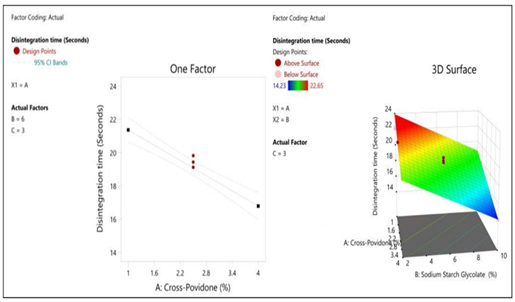
Figure 7: The 2D contour plot and the three-dimensional 3D map shows how the total amounts of super disintegrates (X1) and (X2) affect the disintegration time.
ANOVA for Linear Model
Response 2: Disintegration Time.
| Source | Sum of Squares | df | Mean Square | F-value | p-value | |
| Model | 99.07 | 3 | 33.02 | 56.72 | <0> | significant |
| A-Cross Povidone | 35.78 | 1 | 35.78 | 61.46 | <0> | |
| B-Sodium Starch Glycolate | 50.12 | 1 | 50.12 | 86.09 | <0> | |
| C-Hydroxy propyl Methylcellulose | 0.0055 | 1 | 0.0055 | 0.0095 | 0.9242 | |
| Residual | 6.40 | 11 | 0.5822 | |||
| Lack of fit | 6.16 | 9 | 0.6843 | 5.56 | 0.1616 | Not significant |
| Pure Error | 0.2461 | 2 | 0.1230 | |||
| Cure Total | 105.48 | 14 |
Response 3: Hardness (Kg/cm2)
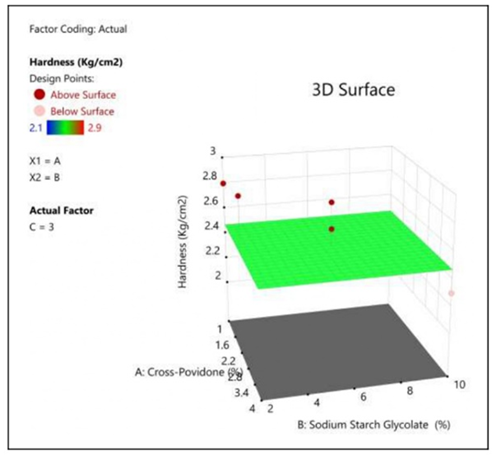
Figure 8: The 2D contour plot and the three-dimensional 3D map shows how the total amounts of super disintegrates (X1) and (X2) affects the Hardness.
ANOVA for Linear Model
Response 3: Hardness
| Source | Sum of Squares | df | Mean Square | F-value | p-value | |
| Model | 0.0000 | 0 | ||||
| Residual | 1.07 | 14 | 0.0764 | |||
| Lack of fit | 0.9427 | 12 | 0.0786 | 1.24 | 0.5307 | Not Significant |
| Pure Error | 0.1267 | 2 | 0.0633 | |||
| Core Total | 1.07 | 14 |
Table 15: Pre compressional data of optimized batch (FDT 16).
| S. No. | Pre-compressional Evaluation Parameter | Results |
| 1. | Bulk Density | 0.48 g/cm2 |
| 2. | Tapped Density | 0.62 g/cm2 |
| 3. | Hausner’s Ratio | 1.29 |
| 4. | Carss Index | 22.58% |
| 5. | Angle of Repose | ~30° to 35° |
Table 16: Post compressional data of optimized batch (FDT 16).
| S. No. | Post-Compression Evaluation Parameter | Results |
| 1. | Thickness | 2.98±0.2mm |
| 2. | Hardness | 2.357 kg/cm2 |
| 3. | Disintegration Time | 13.8 seconds |
| 4. | Friability | 0.49 % |
| 5. | % Drug Release | 97.981 % |
Table 17: In vitro drug release for optimized batch (FDT 1).
| Formulation Batch | 2.5 min | 5 min | 10 min | 15 min | 20 min |
| FDT 1 | 42.17 | 59.89 | 77.89 | 89.69 | 98.56 |
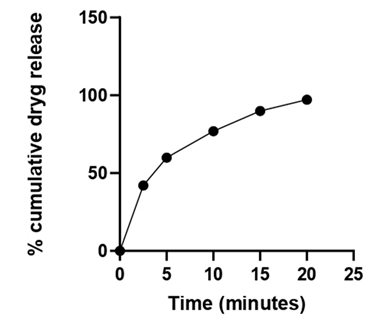
Figure 9: Percent drug release of batch FDT 1 (Optimized Batch).
The obtained Percent drug release of batch ODT 16 (optimized batch) has 97.21% of drug release in 20 minutes, and the optimized batch was evaluated for all the pre and post compressional parameters required for quality control of dosage form and the obtained results was found between satisfied range.
Stability Studies
The stability studies of fast-dissolving tablets of Glipizide at a 100 mg dose indicated that the best formulation remained stable even after storing at 40±20°C / 75±5% RH for 3 months. The tablets were visually examined for any physical changes, evaluated for drug content, and in vitro drug release at monthly intervals. The results showed that the formulation maintained its drug release profile within the specified limits, demonstrating stability over the study period.
Conclusion
This study has convincingly shown the effectiveness of using a Box–Behnken design to improve the of fast-dissolving glipizide tablets through direct compression. A thorough investigation into the combined effects of super-disintegrants specifically cross-povidone and sodium starch glycolate alongside hydroxypropyl methylcellulose as the binder, resulted in a formulation that features significantly quick disintegration and an enhanced drug release profile. This innovative dosage form effectively addresses important issues in managing Type 2 diabetes, specifically helping patients with dysphagia who require a treatment that is easy to take. The rapid dissolution of the tablets, which eliminates the need for water, greatly enhances patient adherence and treatment effectiveness. Given the ongoing global health challenges posed by diabetes, these results represent a significant step forward in patient-focused drug delivery systems, highlighting the value of the Box–Behnken design in pharmaceutical development and paving the way for future advancements in the treatment of chronic metabolic conditions.
Declarations
Acknowledgement
I want to express my sincere thanks to IPS Academy College of Pharmacy for providing the vital resources and facilities that significantly aided my academic path. I also truly appreciate the invaluable support and engaging conversations I had with my colleagues and peers. Their encouragement, helpful feedback, and emotional support were crucial to achieving success in this project. I am genuinely grateful for their contributions, which have been essential to completing this work.
Conflict of Interest
The authors declare no conflict of interest.
References
- Parasuraman S, Thinagaran JN. (2023). Evaluation of the Antidiabetic Activity of Hesperidin on Streptozotocin-Induced Diabetes Mellitus in Swiss Albino Mice. Free Radicals and Antioxidants. 13(1):46-49.
Publisher | Google Scholor - Tiwary N, Sharma N, Singh S, Behl T, Zahoor I. (2024). Understanding The Pharmacological and Nanotechnological Facets of Dipeptidyl Peptidase-4 Inhibitors in Type II Diabetes Mellitus: A Paradigm in Therapeutics. Bionanoscience. 14(1):211-229.
Publisher | Google Scholor - Li Z, Mei S, Dong Y, She F, Li Y, et al. (2020). Functional Nanofibrous Biomaterials of Tailored Structures for Drug Delivery a Critical Review. Pharmaceutics. 12(6):522.
Publisher | Google Scholor - El-Dakroury WA, Zewail MB, Amin MM. (2023). Design, Optimization, And In-Vivo Performance of Glipizide-Loaded O-Carboxymethyl Chitosan Nanoparticles in Insulin Resistant/Type 2 Diabetic Rat Model. Journal of Drug Delivery Science and Technology. 79:104040.
Publisher | Google Scholor - Omar SM, Abdalla FI, Abdelgawad NM. (2018). Preparation and Optimization of Fast-Disintegrating Tablet Containing Naratriptan Hydrochloride Using D-Optimal Mixture Design. AAPS Pharmscitech. 19:2472-2487.
Publisher | Google Scholor - Alburyhi MM, Saif AA, Noman MA, Yahya TA, Al-Ghorafi MA. (2023). Formulation and Evaluation of Drotaverine Orally Disintegrating Tablets. World Journal of Pharmaceutical Research. 12(18):66-79.
Publisher | Google Scholor - Singh A, Chauhan CS. (2024). Formulation and Optimization of Valganciclovir-Loaded Nano Sponges. International Journal of Drug Delivery Technology. 14(1):1-8.
Publisher | Google Scholor - Roy H, Nandi S. Pavani U, Lakshmi U, Reddy TS, Gayatri DV. (2020). Optimization and Quality by Design Approach for Iroxicam Fast Dissolving Tablet Formulations Using Box-Behnken Design. Current Drug Therapy. 15(2):152-165.
Publisher | Google Scholor - Sharma S, Chauhan S. (2021). A Review: An Overview of Natural Super Disintegrants. Research Journal of Topical and Cosmetic Sciences. 12(1):13-24.
Publisher | Google Scholor - Butola M, Bhatt V, Nainwal N, Jakhmola V, Dobhal K, et al. (2023). Preparation and Evaluation of Polymer Fused Metformin Hydrochloride Sustained Release Tablet. Indian Journal of Pharmaceutical Education and Research. 57(3):711-717.
Publisher | Google Scholor - Kakad SB, Rachh PR. (2022). Effect of Hydrophilic Polymer and Binder on Drug Release of Metformin Hcl Sustained Release Tablet. International Journal of Health Sciences. 4:3343-6635.
Publisher | Google Scholor - Tomar A, Wadhwani S, Bhadauria RS. (2019). A Preformulation Study of Pure Metformin Hcl. International Journal of Pharmaceutical Erudition. 8(4):1-9.
Publisher | Google Scholor - Tyagia L. (2024). A Comprehensive Review on Superdisintegrants: Accelerating Dissolution for Enhanced Drug Performance: International Journal of Medical Science. 34-49.
Publisher | Google Scholor - Othman AM, Alburyhi MM, Al-Hadad GH. (2024). Formulation and Evaluation of Captopril Mouth Dissolving Tablets. European Journal of Pharmaceutical and Medical Research. 11(1):18-28.
Publisher | Google Scholor - Kusuma AN, Kumar RS. (2024). Optimization Of Fast-Dissolving Tablets of Carvedilol Using 23 Factorial Designs. International Journal of Applied Pharmaceutics. 1:98-107.
Publisher | Google Scholor - Gupta A, Yadav A, Jain DK. (2024). Fast Dissolving Tablets for The Treatment of Acute and Chronic Diseases. International Journal of Pharmaceutical Drug Design. 1(2):1-8.
Publisher | Google Scholor - Turanli Y, Birer M, Birer YT, Uyar R, Dikmen BY, et al. (2024). Oral Fast-Dissolving Risperidone Loaded Electrospun Nanofiber Drug Delivery Systems for Antipsychotic Therapy. Journal of Drug Delivery Science and Technology. 92:1-9.
Publisher | Google Scholor - Alburyhi MM, Noman MA, Saif AA, Salim YA, Abdullah JH. (2024). Formulation and Evaluation of Domperidone Orodispersible Tablets. World Journal of Pharmacy and Pharmaceutical Sciences. 13(3):49-68.
Publisher | Google Scholor - Chatterjee A, Gupta R. (2023). Formulation and Evaluation of Fast Dissolving Tablets of Piroxicam Using Various Concentration of Different Super Disintegrating Agents. International Journal of Health Advancement and Clinical Research. 1(4):90-97.
Publisher | Google Scholor - Akdag Y, Gulsun T, Izat N, Cetin M, Oner L, et al. (2020). Evaluation of Preparation Methods for Orally Disintegrating Tablets. Medicine. 9(1):265-269.
Publisher | Google Scholor - Akdag Y, Gulsun T, Izat N, Cetin M, Oner L, et al. (2020). Characterization and Comparison of Deferasirox Fast Disintegrating Tablets Prepared by Direct Compression and Lyophilization Methods. Journal of Drug Delivery Science and Technology. 57:101760-101772.
Publisher | Google Scholor - Kumar N, Pahuja S. (2019). Dispersible Tablets: An Overview. Journal of Medical Pharmaceutical and Allied Sciences. 8:2183-2199.
Publisher | Google Scholor - Gandhi L, Akhtar S. (2019). Comparative Study on Effect of Natural and Synthetic Superdisintegrants in The Formulation of Orodispersible Tablets. Journal of Drug Delivery and Therapeutics. 9(2):507-513.
Publisher | Google Scholor













