

Case Report
Case report on Kartagener Syndrome, supervised by Professor Cintia Horta Rezende Goulart and co-supervised by Doctor Paula Renata Ferreira Viana
- Catarine Reis De Sousa Prata 1
- Cintia Horta Rezende 2*
- Daniella Araujo Ferreira 3
- Paula Renata Ferreira Viana 4
1Medical student at the Faculty of Medical Sciences of Minas Gerais, Belo Horizonte, MG-Brazil.
2Doctor from the Federal University of Minas Gerais, MG-Brazil.
*Corresponding Author: Cintia Horta Rezende
Citation: C.R.D. Prata, Cintia H. Rezende, Daniella A. Ferreira, P.R.F. Viana. (2024). Case report on Kartagener Syndrome, supervised by Professor Cintia Horta Rezende Goulart and co-supervised by Doctor Paula Renata Ferreira Viana. Journal of Neuroscience and Neurological Research. BioRes Scientia Publishers. 3(1):1-4. DOI: 10.59657/2837-4843.brs.24.017
Copyright: © 2024 Cintia Horta Rezende, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: May 07, 2024 | Accepted: May 28, 2024 | Published: June 04, 2024
Abstract
Introduction: Kartagener syndrome is a rare and hereditary disease, defined by the clinical triad: chronic pansinusitis, bronchiectasis and situs inversus. There is characteristically impairment of ciliary motility.
Case presentation: A 33-year-old man, born in Belo Horizonte, in the Southeast region of Brazil, diabetic and asthmatic, presented to the Hospital Universitário Ciências Médicas, also located in Belo Horizonte, with an exacerbation of recurrent episodes of productive cough, fever, dyspnea and asthenia. Clinical and imaging findings revealed acute pneumonia, bronchiectasis, dextrocardia and situs inversus. He was treated with oral antibiotics, bronchodilators, corticosteroids, mucolytics and respiratory physiotherapy, with clinical improvement. He was discharged from hospital for outpatient clinical control.
Conclusion: Patients with Kartagener syndrome present with chronic recurrent sinopulmonary infections. The objective of treatment should be the prevention of chronic lung lesions and bronchiectasis, with the two pillars of respiratory treatment being antibiotic therapy and respiratory physiotherapy. The prognosis is generally considered favorable, and life expectancy is usually normal, with regular clinical monitoring.
Keywords: bronchiectasis; ciliary dyskinesia; sinusitis; situs inversus; kartagener's triad
Introduction
Kartagener Syndrome (KS) is a rare autosomal recessive disease, characterized by a classic triad: chronic pansinusitis, bronchiectasis and situs inversus, with dextrocardia [1]. It was first described by Siewert in 1904, but recognized as a clinical syndrome by Kartagener in 1933 [2]. KS, also known as “Immobile Eyelash Syndrome”, makes up a subgroup of primary ciliary dyskinesias [3,4]. We report here the case of a 33-year-old man with KS.
Case Report
A 33-year-old Caucasian man, born in and living in the city of Belo Horizonte/Minas Gerais, Brazil, was admitted to the Hospital Universitário Ciências Médicas in November 2022. His main complaints were: exacerbation of a chronic productive cough, fever, asthenia, dyspnea, nasal congestion, nausea and vomiting. Diabetic, he had a history of several hospitalizations for treatment of community-acquired pneumonia. Having bronchial asthma diagnosed in childhood, he reported several consultations since then for clinical control. He denied smoking, alcohol consumption and drug allergies. There was no similar illness in his family. On physical examination, he was conscious, oriented, pale (+ / 4 +), hydrated, anicteric, acyanotic, afebrile. He had blood pressure (BP) of 140 x 80 mmHg; heart rate (HR) of 56 beats per minute; arterial oxygen saturation (SATO2) of 92%; respiratory rate (RR) of 18 breaths per minute; capillary perfusion pressure <2>
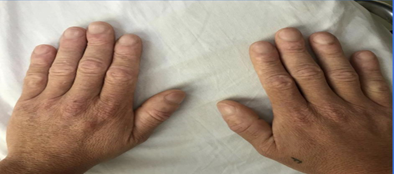
Figure 1:Image of the patient's hands, showing clubbing
Source: personal archive
The chest X-ray revealed the cardiac apex and aortic arch on the right side, and fibrotic bands and bronchiectasis in the lungs (figure 2).
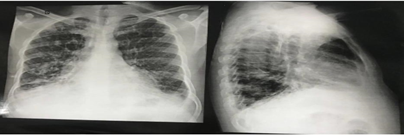
Figure 2:Chest X-ray in posteroanterior and lateral views showing dextrocardia with aortic arch on the right. There are paracardiac and lower lung bronchiectasis with fibrotic bands.
Source: personal archive
Computed tomography (CT) of the chest and abdomen showed dextrocardia, prominent bronchiectatic changes in both lower lung fields, liver and inferior vena cava on the left and spleen on the right, suggestive of situs inversus (figures 3 and 4). Then, the diagnosis of KS was confirmed based on the clinical presentation and the characteristics of the images presented above (figures 1-4). The patient was treated with antibiotics, corticosteroids and bronchodilators administered orally, in addition to respiratory physiotherapy. His clinical symptoms improved and he was then referred for outpatient clinical follow-up with a pulmonologist.
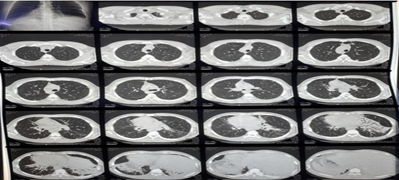
Figure 3: Axial computed tomography of the chest and abdomen showing cystic bronchiectasis in both lungs, situs inversus (liver and inferior vena cava on the left side and spleen on the right side).
Source: personal archive
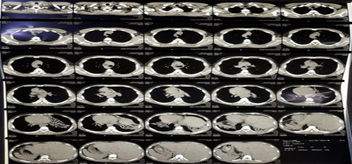
Figure 4:Axial computed tomography of the chest and abdomen showing cystic bronchiectasis in both lungs, situs inversus (liver and inferior vena cava on the left side and spleen on the right side).
Source: personal archive
Discussion
KS is a rare, chronic and progressive autosomal recessive disease, characterized by severe structural or functional impairment of the cilia, with reduced mobility [5]. It is considered a congenital disease, characterized by the classic triad of sinusitis, bronchiectasis and situs inversus [6]. Also known as “Immobile Eyelash Syndrome”, it makes up a subgroup of primary ciliary dyskinesias with mutations in the DNAI1 and DNAH57 genes. Characteristically, there is impairment of sperm tail motility, fallopian tube cilia, respiratory tract cilia, and visceral orientation during embryogenesis, which can lead to male and female infertility, ectopic pregnancy, recurrent sinopulmonary infections and errors of right-left body orientation [3,6,16]. Furthermore, there may be a history of neonatal distress, chronic rhinorrhea and bronchiectasis since childhood1,6. SK can be classified as partial or total. In partial KS, there is dextrocardia, with a change in the position of the heart from left to right. In total KS, in addition to dextrocardia, the liver is located on the left [4].
It is important to highlight that this syndrome occurs in several countries. A study carried out by Guillot (2019) demonstrated a higher prevalence of the syndrome in females - in six of the seven patients evaluated. The diagnosis is based on clinical presentation and imaging tests (such as chest x-rays and chest and abdominal tomography scans) [8]. Treatment is based on the use of bronchodilators, corticosteroids, mucolytics and antibiotics in cases of exacerbation of the disease, which can be associated with both pneumonia and acute sinusitis and otitis [1,9,10,15]. In order to carry out pulmonary rehabilitation, physiotherapeutic exercises must be carried out to promote bronchial hygiene, improve physical conditioning and reduce mucus retention. Respiratory physiotherapy aims to improve symptoms, reduce complications of the underlying lung disease and increase the patient's quality of life [11,16].
Conclusion
Patients with Kartagener Syndrome have chronic recurrent sinopulmonary infections and there is no easy and reliable non-invasive diagnostic test for early diagnosis [12]. The objective of treatment should be the prevention of chronic lung lesions and bronchiectasis [13,14]. The two pillars of respiratory treatment are antibiotic therapy and respiratory physiotherapy. Mucolytic medications, corticosteroids and bronchodilators can be used. The prognosis is generally considered favorable and life expectancy is usually normal, with regular clinical monitoring [10]. Therefore, despite being a rare disease, it is necessary for the healthcare team to be attentive to clinical signs and symptoms to establish a correct and early diagnosis and prevent complications, through regular clinical monitoring and follow-up.
References
- GUIDE T.V. (2022). Kartagener syndrome: a case report. Ibero-American Journal of Humanities, Sciences and Education. São Paulo, 8(5):838-849,.
Publisher | Google Scholor - GOMES, Juliana de Oliveira et al. (2008). Kartagener syndrome. Case report. Brazilian Journal of Clinical Medicine, 210-212
Publisher | Google Scholor - GUILLOT, C.C. et al. (2019). Kartagener syndrome. Cuban Journal of General Integral Medicine, 35(2):1-14.
Publisher | Google Scholor - MEDEIROS, A.S.A. et al. (2020). Kartagener syndrome: limiting aspects to sperm motility and the applicability of assisted fertilization. Research, Society and Development, 9:(9), 1-22.
Publisher | Google Scholor - M.W. Leigh et al. (2009). Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet Med, 11:473-487.
Publisher | Google Scholor - Mishra M et al. (2012). Kartagener's syndrome: A case series. Lung India, 29(4):366-369.
Publisher | Google Scholor - Pederson H, Mygind N. (1976). Absence of axonemal arms in nasal mucosal cilia in Kartagener's syndrome. Nature 262:494-495.
Publisher | Google Scholor - 8SANTANA, Camila P et al. (2004). Kartagener syndrome: case report. Brazilian Journal of Otorhinolaryngology, 857-860.
Publisher | Google Scholor - Homma H et al. (1983). Diffuse panbronchiolitis. A disease of the lung. Chest, 83:63-69.
Publisher | Google Scholor - Oliveira CS et al. (2021). Kartagener syndrome: two cases in the Federal District. Brasília Med, (58):1-6.
Publisher | Google Scholor - VIANA, A.C. et al. (2019). Assessment of lung function, response to exercise and quality of life in a patient with kartagener syndrome – case report. Inspirar Magazine, 19(2):1-15.
Publisher | Google Scholor - RUMMAN N, Jackson C, Collins S, Goggin P, Coles J, Lucas JS. Diagnosis of primary ciliary dyskinesia: potential options for resource-limited countries. Eur Respir Rev, 26(143):160-168.
Publisher | Google Scholor - Brazilian Guidelines for the Management of Chronic Cough. (2006). BRAZILIAN JOURNAL OF PNEUMOLOGY.
Publisher | Google Scholor - Kartagener M. (1933). Zur Pathologie der Bronchiektasien: Bronchiektasien bei situs viscerum invertus. Beitr Klin Tuberk, 83:489-501.
Publisher | Google Scholor - QUIRINO, I. C. P.; CUNHA, R. M.; DEL FIACO, L. G.; MIGUEL, M.; CALVOSO, B. de S.; BUENO, L. C. V.; PANINI, V. de O.; XAVIER, I. M.; CARDOSO, L. L.; GUIMARÃES, T. S.; VARGAS, R. G. Kartagener syndrome: a diagnostic approach, clinical evolution and review. Brazilian Journal of Health Review.
Publisher | Google Scholor - OLIVEIRA, Giovanna; FREITAS, Maria; DARDIS, Maroa; SOUZA, Alysson; SOUSA, Daniela. Aspects and psychosocial impacts of Primary Ciliary Dyskinesia in patients and caregivers. Brazilian Journal of Health Review, Curitiba, 6(4).
Publisher | Google Scholor













