

Case Report
Bone Plasmacytoma in Patients Younger Than 30 Years-Case Report and Review of the Literature
Department of Internal Medicine, Division of Hematology-Oncology, Lebanese American University Medical Center Rizk Hospital. Beirut, Lebanon.
*Corresponding Author: Riad Akoum, Department of Internal Medicine, Division of Hematology-Oncology, Lebanese American University Medical Center Rizk Hospital. Beirut, Lebanon.
Citation: R. Akoum. (2024). Bone plasmacytoma in patients younger than 30 years: Case report and review of the literature. International Clinical and Medical Case Reports, BioRes Scientia Publishers. 3(1):1-7. DOI: 10.59657/2837-5998.brs.24.031
Copyright: © 2024 Riad Akoum, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: December 19, 2023 | Accepted: December 29, 2023 | Published: February 08, 2024
Abstract
This is the case report of a 26-year-old female who was diagnosed with bifocal bone plasmacytoma of the proximal femur and the proximal humerus. The tumour cells showed a plasmablastic morphology. After successful treatment with surgery and radiation therapy, the tumour recurred 10 years later in the left adrenal gland. We present the clinical, radiological and pathological findings and we discuss the differential diagnoses. We have also performed a review of the published cases younger than thirty years old.
Keywords: Bone Plasmacytoma; Young patients; Plasmablastic morphology; Myeloma
Introduction
Solitary plasmacytoma of the bone (SBP) is a rare plasma cell neoplasm without evidence of systemic plasma cell proliferative disease. It accounts for 2% to 5% of all plasma cell neoplasms [1,2,3]. The median age at diagnosis is 55 years, occurring 10 years earlier than multiple myeloma (MM) [4]. SBP has been reported in patient as young as 15 years (5), although reported cases in patients under 30 years of age are rare (Table 1) [6-26]. Males are more frequently affected than females. It occurs mostly in the vertebras and in the skull [2,3]. SBP is considered to be a precursor lesion to MM with conversion rates of 65% to 100% at 15 years. The morphologic spectrum of tumour cells ranges from cells with mature plasmacytic differentiation to large immature cells called plasmablasts. The immuno-phenotype is CD20, CD38+ and CD138+. SBP is a single lytic lesion without systemic marrow plasmacytosis. Multiple solitary plasmacytomas occur in up to 5% of patients and may involve the soft tissues or the bones [20]. Diagnostic criteria include normal bone marrow, normal skeletal survey, normal MRI of spine and pelvis and no related organ impairment. A monoclonal gammopathy may be seen in 24-72% of cases [21]; however, the monoclonal protein should disappear after resection of the lesion. Eighty five percent of SBP develop a local recurrence (12%), a new solitary lesion (15%), or MM (58%) [22]. Dissemination to MM is more commonly seen with primary lesions in the spine than with lesions in peripheral bones (61% vs 26%). Predictors of dissemination are age older than 50 years and the presence of monoclonal component after local treatment [22].
Table 1: Reported cases of bone plasmacytoma in young patients under 30 years of age.
| Author Year | Age | Sex | Location | pathology | Focality | Treatment | Serum paraprotein | Ref |
| Dodge 1964 | 20 25 25 30 | M M M F | Femur Foot & Tibia Sacrum Tibia | - | Monofocal Bifocal Monofocal Monofocal | - - - - | - | [6] |
| Ishida 1995 | 21 | M | Tibia | Mature | Monofocal | - | - | [7] |
| Boos 1997 | 16 | F | L2 | Mature | Monofocal | Surgery + Radiation (30Gy) | - | [8] |
| Mongknosritragoon 1998 | 27 | M | Tibia Rt & Lt | Mature | Bifocal | Radiation (40 Gy) | IgG λ | [9] |
| Bertoni-Salateo 1998 | 17 | F | Tibia | Mature & Plasmablastic | Monofocal | Radiation (36 Gy) | κ | [10] |
| George 2002 | 29 | M | Rib | Mature | Monofocal | Surgery | IgG κ | [11] |
| Gossios 2002 | 18 | M | T12 | Mature | Monofocal | Surgery Chemotherapy | IgG κ | [12] |
| Yamac 2002 | 29 | F | Tibia | Mature | Monofocal | - | IgG κ | [13] |
| Panteli 2002 | 18 | M | T | Mature | Monofocal | Surgery | [14] | |
| Hussein 2003 | 28 | M | Rib Mandible | Mature | Bifocal | Chemotherapy | κ | [15] |
| Panagopoulos 2006 | 27 | M | Clavicle | Matuire | Monofocal | Radiation Chemotherapy | - | [16] |
| Dumesnil 2006 | 14 | F | L | Mature | Monofocal | - | IgG λ | [17] |
| Kumar 2011 | 14 | F | Tibia | Mature | Monofocal | - | [18] | |
| Rago 2010 | 22 | M | Tibia | Plasmablastic | Monofocal | Radiation (40 Gy) | - | [19] |
| Kulbacki 2011 | 20 | M | C1 Rt hip | Mature | Bifocal | Radiation Chemotherapy | - | [20] |
| Pasch 2012 | 21 | M | Iliac crest | Mature | Monofocal | Radiation (50 Gy) | - | [21] |
| Jacob 2014 | 16 | M | Metacarpal bone | Mature | Monofocal | - | IgG κ | [22] |
| Singh 2016 | 25 | M | Skull | Mature | Monofocal | - | - | [23] |
| Bouattour 2018 | 21 | F | Spine | Mature | Monofocal | Surgery | IgA κ | [24] |
| Lee 2020 | 24 | F | Skull Spine | Mature | Multifocal | Chemotherapy | IgG λ | [25] |
| Franck 2021 | 7 | F | Iliac wing | Mature | Monofocal | - | IgG λ | [26] |
Case report
A 26-year-old female, previously healthy, presented in 2009 with one month history of right hip heaviness and pain causing limping. The pain was aggravated by activity, improved by rest and absent at night. The patient denied any fever, chills, night sweats or weight loss. There were no palpable masses, lymphadenopathy, neurological deficiency or skin abnormality. The right hip joint range of motion was considerably decreased. The anteroposterior and lateral radiographs views revealed a radiolucent lesion with bone erosion and irregularity of right hip neck extending to the intertrochanteric area. The MRI revealed a 3.5 x 3 x 2 cm tumor in the right femur neck into the intertrochanteric area with irregular inferior border, iso signal on T1, faint hyper signal on T2 but shining hyper signal on STIR sequence. A biopsy was taken and before the results were finalized, the patient suffered a pathological fracture which required a right total hip replacement. The tumor was composed of a relatively monomorphic population of cells that have an abundant cytoplasm and mitotic activity, only mild pleomorphism and only mild cytological atypia. The cells showed no evidence of rosette formation or gland formation. The tumor cells expressed: strongly CD138, CD38, CD79a, IgG lambda and focally CD56. However, CD5, CD3 were expressed by scattered lymphocytes and CD117 CD99, CD34, CD63, CD45, CD15, CD30, CD20, S100, MPO and Alk were not expressed. Three months later, the patient underwent a total body MRI with background body signal suppression (DWIBS) which revealed a zone of diffusion in bone marrow at the level of the upper left humerus. Biopsies were taken and showed a large number of atypical cells (64%) of variable sizes, sometimes gigantic and strongly positive to immunostaining with CD138 and negative to PAS and CD99. Shortly after this finding the patient experienced a pathological spontaneous fracture which was surgically fixed. Bone marrow examination did not show any morphologic or phenotypic evidence of plasma cell dyscrasia or lymphoma. The serum and urinary protein electrophoresis and immunoelectrophoresis were normal. The complete blood cell count, liver function tests, serum lactate dehydrogenase and beta-2 microglobulin levels, renal function tests and calcium level were normal. The serologic screening assays for HIV and EBV were negative. Based on the imaging studies, the history and the pathological appearance with the presence of plasmablasts, nuclear inclusions (Dutcher bodies) and positive CD138 the patient was diagnosed as having a myelomatous proliferation with plasmablastic component and anaplastic cells and started on chemotherapy using bortezomib and dexametazone. She also received radiotherapy to the two involved region for a total dose of 36 Gy. The chemotherapy has been stopped 8 months later and the patient was followed up every 6 months for 18 months. Then she re-experienced heaviness in the right leg and limping. The imaging studies revealed a hyperintense signal on the pelvic girdle which seemed to arise from the hip joint just next to the radiation field. Further staging work-up including bone scan and PET CT scan were negative. The recurrence has been successfully treated by radiation therapy. The patient has remained in persistent complete remission for 9 years. Then she developed abdominal pain and distention, the PET CT scan revealed the presence of active bulky left adrenal gland, a big intra-abdominal mass causing intestinal obstruction and multiple lytic bone lesions. She underwent a palliative abdominal surgery. The biopsy confirmed the recurrence of plasmablastic plasmacytoma. The bone marrow studies revealed the presence of 10% plsma cells and the serum protein electrophoresis and immunoelectrophoresis showed an IgG lambda monoclonal gammopathy.
In summing up, this is a young female patient found to have two osteolytic lesions showing plasmablastic morphology with normal bone marrow and normal blood chemistry. We discuss in this report the differential diagnoses and the relationship between plasmablastic bone plasmacytoma and plasmablastic lymphoma.
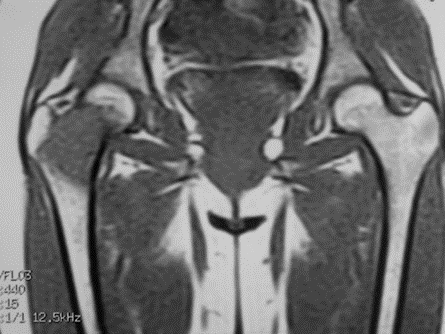
Figure 1: A 4.1 x 3.2 x 2 cm cystic lesion in the neck of the right hip into the intertrochanteric area.
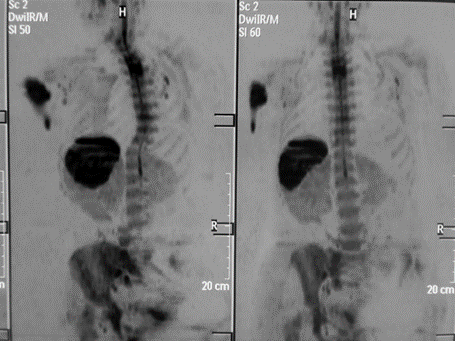
Figure 2: Total body MRI, diffusion in the bone marrow at the level of the upper left humerus.
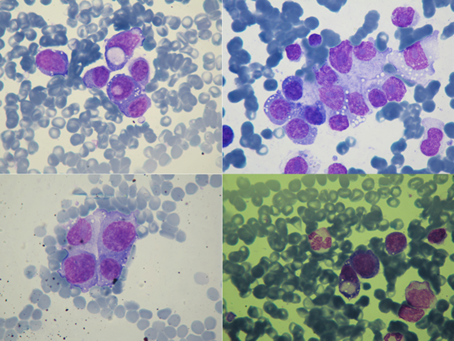
Figure 3: Atypical cells of variable sizes, sometimes gigantic, from the bone marrow of the left humerus.
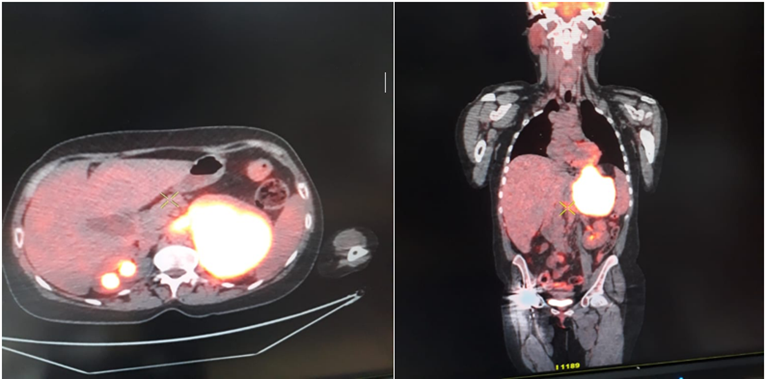
Figure 4: PET CT scan showing the left adrenal recurrence 10 years after remission.
Discussion
The osteolytic, intramedullary lesion extending from the proximal femoral metaphysis through the subchondral bone occurring in this 26-year-old lady suggested differential diagnoses including; giant cell tumor, benign fibrous histiocytoma (BFH), Langerhans cell histiocytoma (LCH), plasmacytoma, lymphoma, osteosarcoma and osseous Ewing sarcoma. Based on the lesion location and the patient age the most likely diagnosis was giant cell tumor of bone, which represents 15% of all bone tumors with a peak incidence between 10 and 45 years and female predilection. Although, pathological fracture may ensue patients with BFH and LCH report pain with no impairment of the joint function. LCH usually occurs in children and adolescents and there is no defined age group for BFH occurrence. Osteosarcoma and Ewing sarcoma were considered less likely because of the absence of periosteal reaction, soft tissue extension and intra-lesion mineralization. Primary lymphoma of the bone has been defined variably as lymphoma arising in the medullary cavity of one or multiple bones without regional lymph node involvement. In SBP There is a male predilection with a median age at presentation of 55 years. Sarcoma and carcinoma markers were negative in histopathology and the cytomorphological studies confirm the plasmablastic nature of this tumor. The full skeletal survey, the MRI and the PET CT scan revealed the involvement of two different and unusual skeletal sites with normal bone marrow and normal serum and urine protein electrophoresis and immunofixation. These characteristics could fit with the diagnosis of high-grade lymphoma or plasmablastic plasmacytoma. The clinical behaviors of these tumors and their treatments are radically different.
Primary bone lymphomas account for approximately 3% to 7% of all primary osseous malignancies and 92% of them are diffuse large B-cell lymphoma. The peak age at presentation is during the fifth decade. Tumors are located in lower extremities in 30% of cases, most frequently in metadiaphysal femur and pelvis. The lesions are purely lytic in 70% of cases without appreciable matrix production. Periosteal reaction is evident in 50% of cases and 20% of patients present with an associated pathological fracture [23]. They are usually CD20 positive and occasionally CD10 positive. Highly aggressive plasma cell MM or SBP may contain a predominance of plasmablasts which can closely resemble the malignant cells of plasmablastic lymphoma and other B-cell lymphoma with predominant plasmablastic features including Alk positive large B-cell lymphoma, HHV8 positive, Castelman disease and lymphoma of pleural effusion [27-32]. In both cases, plasmablasts express weakly the conventional B-cell markers and strongly the post-germinal center B-cell and plasma cell markers: CD138, MUM1, CD38 and CD56. In this case, the tumor cells did not express CD19 and CD45, while focally expressed CD56 and strongly CD38, CD138 and CD79a. The EBV is usually negative in plasmacytoma and positive in 60% of plasmablastic lymphoma. The distinction between plasmablastic lymphoma and plasmablastic myeloma depends heavily on clinical correlation. The presence of two distant lytic bone lesions favors the diagnosis of plasma cell neoplasm. Although MM and SBP may occasionally occur in the setting of HIV positive individuals, they do not have the strong association with AIDS that has been reported in plasmablastic lymphoma.
Although plasmablastic morphology has been reported to confer a poor clinical prognosis in plasma cell tumors [38]. However, the risk stratification relies primarily on clinical staging and cytogenetic abnormalities. It has also been suggested that in immunocompromized patients, plasmablastic tumors may have been driven by EBV to gain the plasmablastic cytomorphologic features and high proliferation index. EBER-positive plasmablastic plasma cell tumors rarely exist in immunocompetent patients [38]. Vega et al [32] found that the only significant difference between MM and PL was the presence of EBV-encoded RNA (EBER) which was positive in all plasmablastic lymphoma cases with or without HIV positive status. They suggested that when considering a single case with differential diagnosis between MM and PL, a positive EBER result makes the diagnosis of MM unlikely. Neither immunostaining for EBV nor in situ hybridization for EBV-encoded RNA was performed in the tumor specimen; however, the serologic studies were negative for IgM and IgG.
Young patients with plasma cell neoplasm, who present with multiple lytic bone lesions but without intervening infiltration of bone marrow, have been identified as having macro-focal MM. They usually are less anemic, with no hypercalcemia or renal failure, normal LDH level and uninvolved immunoglobulins and better response rate to primary treatment and long-term survival. A retrospective study of SEER database [40] collected 1164 patients over a 30-year-period. Only 1.1% was younger than 30 years and had better prognosis with 90% 5-year survival. No significant difference in survival between patients with single or 2 lesions was found; however, patients with more than 2 lesions had poorer prognosis. The use of any local disease control modality, either surgery or radiation therapy was associated with better outcomes. A preceding trauma was identified in a number of reported cases [7, 8, 9, 14, 15, 16, 17, 19] of SBP and given the rarity of such a tumor some authors suggested that trauma may play a role in the pathogenesis by acting as triggering stimulus for plasma cells to proliferate.
CD56 is expressed in NK cells and NK-like T-cells but it is aberrantly expressed in MM (75%) and it can distinguish plasma cells of MM from those of reactive plasmacytosis, MGUS, and lymphoplasmacytic lymphoma. CD56 expression has also been associated with osteolysis and poorer outcome [41]. Progression to multiple myeloma is the most important factor for overall survival in patients with SBP. The role of surgery alone and chemotherapy alone in achieving long term remission remains controversial. Radiation therapy; 30-50 Grays, may achieve 82% complete response rate with acceptable toxicity.
References
- Knowling MA, Harwood AR, Bergsagel DE. (1983). Comparison of extramedullary plasmacytomas with solitary and multiple plasma cell tumors of bone. J Clin Oncol. (4):255-62.
Publisher | Google Scholor - Dimopoulos MA, Moulopoulos LA, Maniatis A, Alexanian R. (2000). Solitary plasmacytoma of bone and asymptomatic multiple myeloma. Blood. 5;96(6):2037-44.
Publisher | Google Scholor - Dimopoulos MA, Hamilos G. (2002). Solitary bone plasmacytoma and extramedullary plasmacytoma. Curr Treat Options Oncol. 3(3):255-9.
Publisher | Google Scholor - Shih LY, Dunn P, Leung WM, Chen WJ, Wang PN. (1995). Localized plasmacytomas in Taiwan: comparison between extramedullary plasmacytoma and solitary plasmacytoma of bone. Br J Cancer. 71(1):128-33.
Publisher | Google Scholor - Pavithran K, Doval DC, Rao CR, Bapsy PP, Anantha N. (1997). Pediatric solitary plasmacytoma. Acta Oncol. 36(1):83-4.
Publisher | Google Scholor - Dodge OG. (1964). BONE TUMOURS IN UGANDA AFRICANS. Br J Cancer. 18:627-33.
Publisher | Google Scholor - Ishida T, Dorfman HD. (1995). Plasma cell myeloma in unusually young patients: a report of two cases and review of the literature. Skeletal Radiol. 24(1):47-51.
Publisher | Google Scholor - Boos N, Goytan M, Fraser R, Aebi M. (1997). Solitary plasma-cell myeloma of the spine in an adolescent. Case report of an unusual presentation. J Bone Joint Surg Br. 79(5):812-4.
Publisher | Google Scholor - Mongkonsritragoon W, Kyle RA, Shreck RR, Greipp PR. (1998). Primary plasmacytoma at the site of exit wounds after electrical injury. Am J Hematol. 58(1):77-9.
Publisher | Google Scholor - Bertoni-Salateo R, de Camargo B, Soares F, Chojniak R, Penna V. (1998). Solitary plasmocytoma of bone in an adolescent. J Pediatr Hematol Oncol. 20(6):574-6.
Publisher | Google Scholor - George SM, Ratnakar KS, Shome DK, Nair R, Al Ajmi A. (2002). Plasmacytoma of the rib in young male. Asian Cardiovasc Thorac Ann. 10(3):282-4.
Publisher | Google Scholor - Gossios K, Argyropoulou M, Stefanaki S, Fotopoulos A, Chrisovitsinos J. (2002). Solitary plasmacytoma of the spine in an adolescent: a case report. Pediatr Radiol. 32(5):366-9.
Publisher | Google Scholor - Yamaç K, Fen T, Sancak A. (2002). Solitary Plasmacytoma of Tibia in a 29-Year-Old Woman. Turk J Haematol. 19(4):485-7.
Publisher | Google Scholor - Panteli K, Tsiara S, Bourantas KL. (2002). Solitary bone plasmacytoma in a young patient. J Exp Clin Cancer Res. 21(1):139-41.
Publisher | Google Scholor - Hussein MA, George R, Rybicki L, Karam MA. (2003). Skeletal trauma preceding the development of plasma cell dyscrasia: eight case reports and review of the literature. Med Oncol.20(4):349-54.
Publisher | Google Scholor - Panagopoulos A, Megas P, Kaisidis A. (2006). Radiotherapy-resistant Solitary Bone Plasmacytoma of the Clavicle. European J Trauma.
Publisher | Google Scholor - Dumesnil C, Schneider P, Dolgopolov I, Radi Set, et al. (2006). Solitary bone plasmocytoma of the spine in an adolescent. Pediatr Blood Cancer. 47(3):335-8.
Publisher | Google Scholor - Kumar P, Sharma SC, Saikia UN, Kumar N et al. (2011). Solitary plasmacytoma of the proximal tibia in an adolescent. Pediatr Blood Cancer. 56(1):158-60.
Publisher | Google Scholor - Rago A, Miraglia A, Mecarocci S, Pisanelli GC, et al. (2010). Solitary plasmacytoma of tibia: a possible correlation with younger age. Leuk Res. 34(8): e181-2.
Publisher | Google Scholor - Kulbacki E, Wang E. (2012). Pathological bone fractures in a 20-year old athletic male with multifocal solitary plasmacytoma of bone. Am J Hematol. 87(6):626-7.
Publisher | Google Scholor - Pasch W, Zhao X, Rezk SA. (2012). Solitary plasmacytoma of the bone involving young individuals, is there a role for preceding trauma? Int J Clin Exp Pathol.5(5):463-7.
Publisher | Google Scholor - Jacob PM, Nair RA, Koshy SM, Kattoor J. (2014). Solitary plasmacytoma of the metacarpal bone in an adolescent. Indian J Pathol Microbiol. 57(2):323-5.
Publisher | Google Scholor - SINGH, Swati et al. (2017). Rare case of solitary plasmacytoma of the skull in a young male patient. S. Afr. J. radiol. 21:1, p. 1-4.
Publisher | Google Scholor - Bouattour N, Hdiji O, Hachicha A. (2020). Vertebral solitary bone plasmacytoma in a young adult with Trisomy 21: A case report. J Spinal Cord Med. 43(6):908-911.
Publisher | Google Scholor - Lee M, Zhang X, Nguyen V. (2020). Multiple Myeloma in a Young Female Presenting with Neurological Symptoms. Case Reports in Hematology.
Publisher | Google Scholor - Frank LG, Bertrand KAJ, Celestin BA. (2021). Plasmacytoma of the iliac bone in a seven-year-old girl: A rare caser with no to ignore. J. Ped Surg case report. 101582.
Publisher | Google Scholor - International Myeloma Working Group. (2003). Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 121(5):749-57.
Publisher | Google Scholor - Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, et al. (2011). The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 12;117(19):5019-32.
Publisher | Google Scholor - Bataille R, Sany J, Serre H. (1981). [Apparently isolated plasmacytoma of bone. Clinical and prognostic data. 114 cases and review of literature]. Nouv Presse Med. 14;10(6):407-11.
Publisher | Google Scholor - Mulligan ME, McRae GA, Murphey MD. (1999). Imaging features of primary lymphoma of bone. AJR Am J Roentgenol. 173(6):1691-7.
Publisher | Google Scholor - Greipp PR, Raymond NM, Kyle RA, O'Fallon WM. (1985). Multiple myeloma: significance of plasmablastic subtype in morphological classification. Blood. 65(2):305-10.
Publisher | Google Scholor - Vega F, Chang CC, Medeiros LJ, Udden MM et al. (2005). Plasmablastic lymphomas and plasmablastic plasma cell myelomas have nearly identical immunophenotypic profiles. Mod Pathol. 18(6):806-15.
Publisher | Google Scholor - Greipp PR, Leong T, Bennett JM, Gaillard JP. (1998). Plasmablastic morphology--an independent prognostic factor with clinical and laboratory correlates: Eastern Cooperative Oncology Group (ECOG) myeloma trial E9486 report by the ECOG Myeloma Laboratory Group. Blood. 1;91(7):2501-7.
Publisher | Google Scholor - Rajkumar SV, Fonseca R, Lacy MQ, Witzig TE. (1999). Plasmablastic morphology is an independent predictor of poor survival after autologous stem-cell transplantation for multiple myeloma. J Clin Oncol. 17(5):1551-7.
Publisher | Google Scholor - Carter A, Hocherman I, Linn S, Cohen Y, Tatarsky I. (1987). Prognostic significance of plasma cell morphology in multiple myeloma. Cancer. 1;60(5):1060-5.
Publisher | Google Scholor - Bartl R, Frisch B, Fateh-Moghadam A, Kettner G et al. (1987). Histologic classification and staging of multiple myeloma. A retrospective and prospective study of 674 cases. Am J Clin Pathol. 87(3):342-55.
Publisher | Google Scholor - Goasguen JE, Zandecki M, Mathiot C, Scheiff JM. (1999). Mature plasma cells as indicator of better prognosis in multiple myeloma. New methodology for the assessment of plasma cell morphology. Leuk Res. 23(12):1133-40.
Publisher | Google Scholor - Chang ST, Liao YL, Lu CL, Chuang SS, Li CY. (2007). Plasmablastic cytomorphologic features in plasma cell neoplasms in immunocompetent patients are significantly associated with EBV. Am J Clin Pathol. 128(2):339-44.
Publisher | Google Scholor - Dimopoulos MA, Pouli A, Anagnostopoulos A, Repoussis P et al. (2006). Macrofocal multiple myeloma in young patients: a distinct entity with favorable prognosis. Leuk Lymphoma. 47(8):1553-6.
Publisher | Google Scholor - Jawad MU, Scully SP. (2009). Skeletal Plasmacytoma: progression of disease and impact of local treatment; an analysis of SEER database. J Hematol Oncol. 24; 2:41.
Publisher | Google Scholor - Ely SA, Knowles DM. (2002). Expression of CD56/neural cell adhesion molecule correlates with the presence of lytic bone lesions in multiple myeloma and distinguishes myeloma from monoclonal gammopathy of undetermined significance and lymphomas with plasmacytoid differentiation. Am J Pathol. 160(4):1293-9.
Publisher | Google Scholor













