

Review Article
Bioinformatics-A New Tool in Dentistry
Homeopathy Specialists best doctor in Nirman Nagar in Jaipur, Shining Bright Academy, India.
*Corresponding Author: Manisha Saxena,Homeopathy Specialists best doctor in Nirman Nagar in Jaipur, Shining Bright Academy, India.
Citation: Manisha Saxena. (2024). Bioinformatics-A New Tool in Dentistry. Dentistry and Oral Health Care, BioRes Scientia Publishers. 3(2):1-9. DOI: 10.59657/2993-0863.brs.24.029
Copyright: © 2024 Manisha Saxena, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: March 18, 2024 | Accepted: March 25, 2024 | Published: March 26, 2024
Abstract
Bioinformatics is a new specialization that focuses on using information science to solve biological problems. It deals with the collection, storage, retrieval and analysis of data from databases. Bioinformatics has supported and promoted healthcare research and taken it to the next level. Bioinformatics can support research in dentistry by understanding the underlying pathways and mechanisms in some oral diseases. It may also help in early prediction and personalized cancer treatment, which may prove beneficial in early detection and accurate cancer treatment. Bioinformatics supports the development of patient care databases and X-ray, CT and MRI image analysis. Diagnostic capabilities also multiply with database management. Saliva omics is a sub-specialization of bioinformatics dealing with the knowledge base of saliva enabling a global examination of saliva-relevant data. The integration of bioinformatics with artificial intelligence and machine learning can lead to enormous positive results in the field of personalized medicine and gene therapy research. This overview will help to understand the tools used in bioinformatics and its role in dentistry.
Keywords: bioinformatics; dentistry; DNA; RNA; covid-19
Introduction
Computing in biology is a boon in healthcare because it improves our understanding and improves our decision-making ability. The COVID-19 epidemic has taught us new ways to deal with biological viruses. Consider a bio emergency in the future, we are prepared to handle it better. The genetic material that contains the nucleic acid is isolated and sequenced. The computer compares this new genome to the existing database, identifying the new virus and revealing its relationship to another virus in its vicinity. The analysis will aid in the development of antiviral therapies that target the proteins that are the building blocks of viruses. Disruption of the protein disrupts the structure and function of the virus [1]. Bioinformatics can be defined as an integrative field of mathematics, computer science and biology to solve biological problems. It uses algorithms and statistics. Bioinformatics incorporates the concepts of machine learning, artificial intelligence, and neural networks to analyze biological data to provide useful results and conclusions [2].
Figure 1: Show different data management in modern biology and medicine.
The Human Genome Project was jointly announced by the Prime Minister of the United Kingdom, Tony Blair, and the President of the United States, Bill Clinton, on 26 June 2000. The human genome is the chemical component of the genetic code of humans, as it contains 300 billion blocks of DNA. With the development of new generation technologies and computers, its use in biology and medicine is possible. Computers, with the help of algorithms, can process raw data and systematically display the results. The combination of biology and informatics created a new field called bioinformatics. The central tenets of life are DNA, RNA and protein. Bioinformatics deals with DNA gene sequences or complete genomes, gene localization, three-dimensional protein structure, amino acid sequence, and protein-nucleic acid interaction. A new branch of "-omics" data streams include: transcriptomics, the pattern of RNA synthesis from DNA; proteomics, distribution of proteins in cells; interactomics, patterns of protein-protein and protein-nucleic acid interactions; and metabolomics, the nature and transport patterns of small molecule transformations through biochemical pathways active in cells [1].
Tools And Techniques in Bioinformatics
Importance of biological databases
A biological database is a computer software or web page designed to update, query, and retrieve information. A huge amount of generated data is stored in online databases. Databases can be categorized as primary and secondary databases. Primary databases include experimental data such as nucleotide sequence, protein sequence, or macromolecular structure. E.g.: Protein databank. Secondary databases include results derived from primary data. Eg: Ensemble [2].
Database-Data storage and retrieval
In bioinformatics, databases are used to store and organize data. Many of these entities collect DNA and RNA sequences from scientific papers and genome projects. The Nucleotide Sequence Database of the European Molecular Biology Laboratory (EMBL-Bank) in the United Kingdom, the DNA Data Bank of Japan (DDBJ) and the National Center for Biotechnology Information (NCBI) GenBank in the United States oversee the International Nucleotide Sequence Database Collaboration (INSDC), which facilitates daily data exchange [1].
Table 1: Different types of databases and their websites.
| Data base | Function | Website |
| Protein Information Resource (PIR) | Protein information | (http://pir.georgetown.edu/), |
| Protein Data Bank (PDB) | Protein | (http://www.pdb.org/), |
| NucleicAcid Data Base (NADB) | Nucleic acid | (http://ndbserver.rutgers. Edu/) |
| Molecular Modeling Data Base (MMDB) | 3D macromolecular structure. | And http://www.ncbi.nlm.nih.gov/Structure/MMDB/mmdb.shtml). |
| Kyoto Encyclopedia of Genes and Genomes (KEGG) | Metabolimic pathway | (http://www.genome.jp/kegg/), |
| Database of Interacting Proteins (DIP) | Protein | (http://dip.doe‑mbi. ucla.edu), |
| PubMed | Bibliographic | http://www.ncbi.nlm.nih.gov/PubMed/ |
| Gene Expression Profiling Interactive Analysis (GEPIA) | ||
| Surveillance, Epidemiology, and End Results (SEER) |
Sequence alignment
DNA is the genetic material that acts as a medium for the transmission of genetic information from one generation to the next. All living things diverge from a common ancestor over time through evolution through changes in their DNA. DNA defines species and individuals, making DNA sequence the basis for researching cell structure and function and decoding life's mysteries [4]. DNA sequencing technologies could assist biologists and healthcare providers in a wide variety of applications, such as molecular cloning, breeding, pathogenic gene searches, and comparative and evolutionary studies. DNA sequencing technologies should ideally be fast, accurate, easy to operate and inexpensive. In 1977, Frederick Sanger developed a DNA sequencing technology that was based on the chain termination method (also known as Sanger sequencing), and Walter Gilbert developed another sequencing technology based on chemical modification of DNA and subsequent cleavage at specific bases [5]. The ability to sequence an organism's DNA is therefore one of the most important and primary requirements in biological research. Sequence alignment (SA) is usually the first step performed in bioinformatics to understand the molecular phylogeny of an unknown sequence. This is done by aligning the unknown sequence with one or more known database sequences to predict common parts, as residues fulfil functional and structural roles that tend to be conserved by natural selection during evolution. Sequence alignment is a method of identifying homologous sequences. The sequences can be nucleotide sequences (DNA or RNA) or amino acid sequences (proteins). The rearrangement process may introduce one or more gaps or gaps in alignment. The gap indicates the possible loss or gain of the remainder; thus, evolutionary insertion or deletion (indel), translocation and inversion events can be observed in SA. This is done by aligning the unknown sequence with one or more known sequences to predict common parts. Aligned sequences of nucleotide or amino acid residues are typically represented as rows in a matrix [6].
Alignment types
Sequence alignment is the comparison of residues between sequences. There are two types of sequence alignment, pairwise sequence alignment (PSA) and multiple sequence alignment (MSA). PSA considers two sequences at a time, while MSA aligns more than two related sequences. MSA is more advantageous than PSA because it takes into account more members of the sequence family and thus provides more biological information. Proteins are key biological molecules that carry structural and functional information, and therefore the sequence arrangement at the amino acid level is more important [2].
Alignment methods
Global alignments are performed on sequences of similar length, and alignments must be performed along their entire length. Local comparison is performed to identify local similar regions between sequences. When there is a large difference in the lengths of the sequences to be compared, a local comparison is generally performed, as shown in Fig.2.
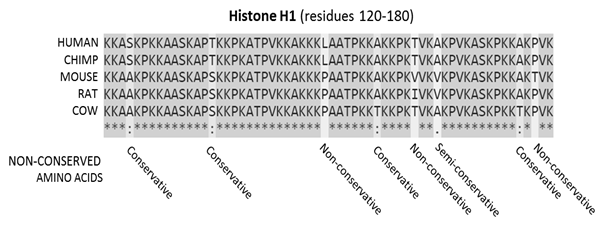
Figure 2: Sequence alignment of a mammalian histone protein
Sequence alignment, produced by ClustalO, of mammalian histone proteins.
The sequences are amino acids for residues 120-180 of the protein. Residues that are conserved across all sequences are highlighted in gray. Below the protein sequences is a key indicating conserved sequence (*), conservative mutations (:), semi-conservative mutations (.) and non-conservative mutations ().
Interpretation
If two sequences in an alignment share a common ancestor, mismatches can be interpreted as point mutations and gaps as indels (ie, insertion or deletion mutations) introduced in one or both lineages at the time they diverged from each other. In a sequence, protein alignment, the degree of similarity between amino acids that occupy a particular position in the sequence can be interpreted as a rough measure of how conserved a particular region or sequence motif is between lineages. The FASTA format is a textual format for representing either nucleotide sequences or amino acid (protein) sequences in which nucleotides or amino acids are represented by single-letter codes and compared to an existing database. Clustal Omega is a multiple sequence alignment program for aligning three or more sequences together in a computationally efficient and accurate manner. Produces a biologically meaningful multiple sequence alignment of divergent sequences [7].
Tools for searching similar sequences in the database
Searching a sequence database for sequences that are similar to a queried sequence is the most common type of database similarity search. The first fast search method was FASTA, which found short common patterns in query and database sequences and combined them into alignments. The Basic Local Alignment Search Tool (BLAST) is similar to FASTA, but is gaining popularity due to its ability to search for rarer and more significant sequences. The PAM250 Scoring Matrix is a scoring matrix based on an evolutionary model that predicts the type of amino acid changes over a long period of time. PSI‑BLAST and PHI‑BLAST are iterative search methods that improve the detection speed of BLAST and FASTA. Pfam and Simple Modular Architecture Research Tool (SMART) are used for protein domain family analysis. Phylogenetic Analysis Using Parsimony (PAUP) and Phylogenetic Inference Package (PHYIP) are software packages available for phylogenetic analysis of macromolecular sequences. The most well-known viewer of macromolecular structures is the RasMol program [10].
Phylogenetic analysis
Phylogenetics is useful for understanding the way species have evolved along with the differences and similarities with their ancestors. Phylogenetics reveals the origin and spread of the human immunodeficiency virus (HIV) and the origin and subsequent evolution of the severe acute respiratory syndrome (SARS) and COVID-19 viruses. This in turn leads us to develop suitable vaccines. Initially, the parameters for evaluating similarities and variations between species were morphological characters, but now they have been replaced in phylogenetics by deoxyribonucleic acid (DNA), ribonucleic acid (RNA), and protein sequence [8]. A phylogenetic tree, also known as a phylogeny, is a diagram that shows the lines of evolutionary descent of different species, organisms, or genes from a common ancestor. It can be rooted or unrooted, bifurcated or branched, circular or rectangular, as shown in Figure 3. A clade is a part of a phylogeny that includes an ancestral line and all descendants of that ancestor [9]. A phylogenetic tree is created as a result of sequence analysis performed on DNA or RNA strands. Various string processing algorithms are available that can rapidly analyze these DNA and RNA sequences and create a phylogeny of sequences or species based on their similarities and dissimilarities. Comparing a given set of sequences with a sequence stored in a database will provide accurate information. This requires comparing multiple sequences by dynamic programming. Sequence comparisons reveal patterns of shared history between species, aiding in the prediction of ancestral states. Phylogenetics is useful for understanding the way species have evolved along with the differences and similarities with their ancestors. Phylogenetics reveals the origin and spread of the human immunodeficiency virus (HIV) and the origin and subsequent evolution of the severe acute respiratory syndrome (SARS) and COVID-19 viruses. This in turn leads us to develop suitable vaccines. Initially, the parameters for evaluating similarities and variations between species were morphological characters, but now they have been replaced in phylogenetics by deoxyribonucleic acid (DNA), ribonucleic acid (RNA), and protein sequence [8]. A phylogenetic tree, also known as a phylogeny, is a diagram that shows the lines of evolutionary descent of different species, organisms, or genes from a common ancestor. It can be rooted or unrooted, bifurcated or branched, circular or rectangular, as shown in Figure 3. A clade is a part of a phylogeny that includes an ancestral line and all descendants of that ancestor [9]. A phylogenetic tree is created as a result of sequence analysis performed on DNA or RNA strands. Various string processing algorithms are available that can rapidly analyze these DNA and RNA sequences and create a phylogeny of sequences or species based on their similarities and dissimilarities. Comparing a given set of sequences with a sequence stored in a database will provide accurate information. This requires comparing multiple sequences by dynamic programming. Sequence comparisons reveal patterns of shared history between species, aiding in the prediction of ancestral states. A phylogenetic tree shows relationships and provides a visual representation of the estimated branching order of taxonomic groups. It can be used to determine the relationships between a collection of viruses, bacteria, animal species, or other operational taxonomic units. They are also important in identifying viral subtypes and new recombinant subtypes arising from combinations of known subtypes. Factors such as population distribution, migration, and changing population size affect the tree. Forces such as recombination, mutation, and selection affect the genetic data of a given tree [2].
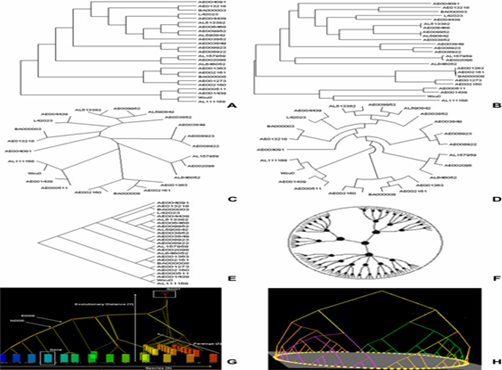
Figure 3: Different types of phylogenetic tree.
A) Example of a cladogram representation: a branching diagram assumed to be an estimate of phylogeny. B) Example of a phylogram. A phylogram differs from a cladogram in that branch lengths are proportional to the amount of inferred evolutionary change. C) Example of an unrooted cladogram. An unrooted tree can be rooted on any of its branches, so many rooted trees can be derived from one unrooted tree. D) Example of a circular cladogram. These types of layouts place nodes in concentric rings around a center. E) Example of a skewed cladogram. The slanted version of the rectangular layout remains just as informative and effective. F) Example of a hyperbolic tree. G) 3D Trees by 3DPE (3D Phylogeny Explorer). H) 3D tree visualized by Arena3D visualization tool.
Prediction of protein structure and function
Protein structure prediction is the derivation of the three-dimensional structure of a protein from its amino acid sequence – that is, the prediction of its secondary and tertiary structure from its primary structure. Protein prediction is essential in drug and vaccine design.
The initial step in protein structure prediction is to confirm whether a protein sequence or part of a protein sequence has any similarities to sequences of known structures present in the Protein Data Bank (PDB) [10].
The Protein Databank is the only worldwide archive of primary structural data of biological macromolecules. Many secondary sources of information are derived from PNR data [11]. Structure prediction is often divided into three areas: ab initio prediction, fold recognition, and homology modeling. The distinction between these areas is usually based on the extent to which information in sequence and structural databases is used in model construction. Ab initio prediction is to predict the structure of a protein entirely based on the laws of physics and chemistry in its purest form and does not rely on information in databases. The term ab initio or de novo is also used for protein structure prediction for which there is no similar structure in the protein database (PDB), but where local sequence and structural relationships involving short protein fragments, as well as secondary structure prediction, are incorporated into the prediction process Recognition of folds, often referred to as "threading", corresponds to the case where one or more structures (templates) similar to a given target sequence exist in the PDB, but cannot be easily identified. The main challenge here is to find the best set of templates, but in general it will be difficult to create an accurate model. At the current stage of the technology, the most accurate models are obtained when a single template can be found in the PNR that has a high level of sequence similarity to the target protein. The process of constructing a model from such a template is called homology or comparative modeling [12].
Internet-based computer systems for protein structure prediction
Access to a computing system designed for bioinformatics analysis is required. For example,a user account on a computer system with a Unix-flavored operating system (e.g., Solaris,Linux or IRIX), sufficient memory, disk space, and applications are required (including an editor, a multiple sequence alignment program, a sequence similarity search program, and access to current biological sequence, structure, and bibliographic databases). . Such facilities are available to registered users of the Human Genome Mapping UK Center (HGMP-RC) [10].
Metabolic pathway
A metabolic pathway is a network of related chemical reactions catalyzed by enzymes that together produce or degrade one or more metabolites. The reconstruction of metabolic networks (grooves) plays an important role in the study of biological systems. Along with other types of biological networks, metabolic pathways can help interpret genotype-phenotype relationships and elucidate the fundamental mechanisms underlying cellular physiology. Most known metabolic pathways are stored in pathway databases such as the Kyoto Encyclopedia of Genes and Genomes (KEGG). High-quality manual annotations of metabolic pathways are a valuable resource for the study of metabolism, but represent only a small fraction of pathways in most organisms. Therefore, the automatic computational reconstruction of metabolic pathways is an important problem to be addressed in bioinformatics and computational biology. The most common approach for constructing metabolic pathways is based on mapping a group of gene and protein sequences of an organism to known reference pathways by sequence homology. The corresponding reference metabolic pathways serve as templates for the placement of genes and proteins (eg enzymes) to construct the metabolic pathways in which they participate [13].
Integration of bioinformatics in dentistry
Oral cancer detection
Cancer is one of the leading causes of death worldwide and will cause approximately 10 million deaths in 2020, according to the World Health Organization. [14] Unfortunately, more than 50% of oral cancer patients show evidence of regional lymph node spread and metastasis at the time of diagnosis, and approximately two-thirds of patients have overt symptoms at presentation, which is a negative prognostic indicator.
Microarray technology
Currently, microarray technology has been widely used in cancer research, diagnosis, and tumor classification for more than a decade. The microarray technique is adopted due to limitations with conventional cancer gene screening techniques, which are mainly time-consuming and cost-inefficient. One of the first applications of microarray was to identify differences in gene expression between normal and cancer cells. DNA microarray analysis includes the use of an oligonucleotide chip, a cDNA chip, and a genomic chip. Oligonucleotide microarrays are used for gene expression studies, single nucleotide polymorphism (SNP), mutational and genotyping analyses, and cDNA microarrays are typically used for gene expression analysis. Microarrays are widely used to understand the genetic and epigenetic makeup of cancer cells. They are also used to decipher the signaling pathways of cancer-relevant transcription factors, which have advanced the scientific understanding of how cancer-relevant transcription factors control gene networks and ultimately cancer development. Microarray technology also makes it possible to examine the state of the cell on a molecular scale and is very crucial in future cancer diagnostics, as traditional methods cannot distinguish between morphologically similar but molecularly different tumors. Molecular differences significantly influence the clinical course of the disease. To date, microarrays are a major tool for global gene expression research for all aspects of human disease and biomedical research [1].
Application of microarrays in oral pathology
In the oral cavity, several conditions, including oral cancer and precancer, have been found to have some genetic basis. Currently, several precancerous conditions and lesions have been identified, although only a small proportion of them progress to malignancy. However, there is no single method by which to predict the behavior of these lesions. It is understood that genetic changes take place long before morphological changes occur in the oral cavity. Microarrays may be able to identify those genetic changes that are more likely to determine the progression of a premalignant lesion to overt malignancy. Targets for drug discovery can also be explored after identifying a gene that plays a significant role in a particular disease. For example, if we compare normal cells with cancer cells and discover a gene that is turned on in a particular type of cancer, we may be able to target that gene in cancer therapy. Today, therapies that directly target dysfunctional genes are already being used and showing exceptional results. Microarrays are commonly used to detect viruses and other pathogens from blood samples. More recently, they have been used to identify heritable markers and have therefore been used as a genotyping tool [3]. Pre-cancers and early stages of oral cancer cannot be adequately identified by visual inspection alone and can easily be missed and neglected, even by highly trained and experienced professionals. A method of detection in the early, curable stages is therefore essential and may lead to a reduction of the currently unacceptably high morbidity and mortality from oral cavity cancer. The development of the OralCDx® system has contributed significantly to eliminating this deficiency. A critical component of OralCDx® is the use of computer-aided image analysis of an oral brush biopsy sample. Applications of the new non-algorithmic neural network computers that were developed in the late 1980s for missile defense. In recent years, neural networks have been successfully applied to several medical diagnostic procedures, including cervical smear screening and the interpretation of digital radiological images such as chest X-rays and mammograms. The OralCDx® neural network helps to find potentially abnormal cells in oral brush biopsy samples, which are then interpreted by a pathologist. Identifying these abnormal cells is laborious, tedious, and time-consuming; more importantly, these abnormalities are also easily missed [15].
Patient care database
Patient care databases are online repositories including data related to patient diagnosis, procedures, drug prescription, etc. Rare case information can be stored and easily accessed. Machine learning algorithms applied to the acquired data can be used to provide appropriate and effective treatment at minimal cost. A personalized well-designed hospital database can be used to collect up-to-date information from the visiting patient. This makes relevant information available at the click of a button. If the patient needs the services of health care providers in different hospitals, an exchange of information is possible. It is an important tool for monitoring and improving health services. May also assist with documentation and invoicing. Reducing paperwork and administrative staff can reduce the cost of running a healthcare facility. Health database organizations (HDOs) have opted for two critical dimensions of databases: comprehensiveness and inclusiveness. Comprehensiveness describes the completeness of patient care event records and information relevant to the individual patient. Inclusiveness refers to which populations in a geographic area are included in the database. The more inclusive a database is, the closer it is to covering 100 percent of the population its developers intend to include [16].
Image analysis
In addition, computational analysis of medical images can enable the discovery of disease patterns and correlations between cohorts of patients with the same disease, thus suggesting common causes or providing useful information for better therapies and treatments. Machine learning and deep learning applied especially to medical images have produced new, unprecedented results that may pave the way to advanced frontiers of medical discovery. To name a few, these imaging modalities include X-ray radiography, magnetic resonance imaging (MRI), positron emission tomography (PET), computed tomography (CT), and single-photon emission computed tomography (SPECT). Since 2010, new computational analyzes of medical images have been made possible through machine learning. It provides highly accurate, time and effort saving and cheaper diagnoses that can be used in the medical imaging profession [17].
Salivaomics Knowledge Base
Saliva (oral fluid) is an emerging biofluid for non-invasive diagnostics used in the detection of human diseases. The need to advance saliva research is strongly emphasized by the National Institute for Dental and Craniofacial Research. (NIDCR) As a computational and informatics infrastructure that can enable global exploration and exploitation, the Salivaomics Knowledge Base (SKB) is being created by combining (1) salivary biomarker discovery and validation resources at UCLA with (2) ontology resources developed by OBO Foundry (Open Biomedical Ontologies) [6], including the new Saliva Ontology (SALO), which is described in this communication. The Salivaomics Knowledge Base (SKB; http://www.skb.ucla.edu/) is a data repository, management system, and web resource created to support human salivary proteomics, transcriptomics, miRNA, metabolomics, and microbiome research. SKB will provide the first web resource dedicated to salivary omics studies and will contain the data and information needed to explore the biology, diagnostic potential, pharmacoproteomic and pharmaco-genomics of human saliva. It has an efficient information retrieval system and carefully designed data format and uses an open data model. Figure 1 shows a three-layer service-oriented SKB architecture with a data layer, an ontology layer, and an interface layer [18].
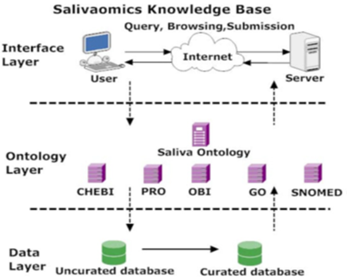
Figure 4: SKB architecture.
SKB is based on a three-tier architecture. Data sources are stored in the data layer. In the ontology layer, data elements from these data sources are mapped to nodes in controlled vocabularies. The interface layer allows querying, browsing, and sending. For example, the interface layer receives user requests, sends queries to the data layer through the ontology layer, and retrieves the corresponding results. These three layers connect data from consumers (users who make queries) to data providers (data sources) through ontologies.
Conclusion
Research in the dental sciences can be metamorphosed by the development of high-throughput techniques and the generation of large amounts of data. Detailed study of molecular sequences and structures can help in understanding genotypes and their clinical phenotypes. The future of dentistry with bioinformatics shows the analysis of clinical data based on genomic information that will increase our understanding of the underlying mechanisms. With this insight, we can change the current practice of dentistry over time, including the diagnosis, therapy, and prognosis of common oral diseases and disorders. This new approach to dentistry will be known at a molecular level that is lacking in today's practice. Genomics and proteomics hold the promise of changing the practice of dentistry and their potential to identify risk factors and therapeutic targets in oncology. Theoretical disciplines such as bioinformatics and data mining will greatly help us understand this complex picture. Saliva Ontology is an ongoing research initiative. The ontology will be used to facilitate the retrieval and integration of salivaomics data across different research areas, along with data analysis and data mining. The current state of the art in genomics and proteomics and theoretical disciplines suggests that an appropriate combination of experimental and theoretical results obtained by different methods will soon become the gold standard for the study of oral cavity diseases.
References
-
-->










