

Case Report
A Hidden Diagnosis Revealed: Primary Hyperoxaluria Unmasked by Bone Marrow Biopsy in a Pancytopenic Patient with End-Stage Renal Disease
1 Section of Hematology & Transfusion Medicine, Department of Pathology and Laboratory Medicine, Aga Khan University, Pakistan.
2 Associate Professor, Section of Hematology & Transfusion Medicine, Department of Pathology and Laboratory Medicine, Aga Khan University, Pakistan.
*Corresponding Author: Muhammad Shayan Ashfaq, Section of Hematology & Transfusion Medicine, Department of Pathology and Laboratory Medicine, Aga Khan University, Pakistan.
Citation: Muhammad S. Ashfaq, Minhas K., Brohi S. (2026). A Hidden Diagnosis Revealed: Primary Hyperoxaluria Unmasked by Bone Marrow Biopsy in a Pancytopenic Patient with End-Stage Renal Disease, Clinical Case Reports and Studies, BioRes Scientia Publishers. 12(3):1-4. DOI: 10.59657/2837-2565.brs.26.313
Copyright: © 2026 Muhammad Shayan Ashfaq, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: March 03, 2026 | Accepted: March 17, 2026 | Published: March 24, 2026
Abstract
The glyoxylate pathway is impacted by primary hyperoxaluria (PH), a rare genetic metabolic disorder that causes an excess of oxalate to be produced. This oxalate builds up in different organs as calcium oxalate. Later on, the crystals may also deposit in non-renal tissues like bone marrow, but the kidneys are usually the first and most severely affected, resulting in progressive kidney damage and recurrent kidney stones. We describe a case of a male patient, age 22, who had end-stage renal disease (ESRD), was receiving twice-weekly hemodialysis, and was the subject of a kidney transplant evaluation. He had had two nephrolithotomy surgeries in the past and had a history of kidney stones that started to occur frequently in early childhood. He eventually developed ESRD that required dialysis as his renal function steadily deteriorated. Unknown cytopenia during his transplant evaluation led to a bone-marrow test. The biopsy showed needle-shaped birefringent crystals encircled by foreign-body giant cells, focal fibrosis, and marrow that was normocellular to mildly hypercellular, with many osteoblasts and osteoclasts along bony trabeculae—findings typical of calcium-oxalate deposition (bone-marrow oxalosis). By connecting these histologic characteristics to the patient's lifetime history of renal failure and nephrolithiasis, the diagnosis of Primary Hyperoxaluria with bone-marrow oxalosis was validated. In patients with ESRD and unexplained cytopenias, this case demonstrates a rare manifestation of PH and the diagnostic utility of bone-marrow evaluation, especially when early-onset nephrolithiasis has been a feature of the patient's clinical trajectory.
Keywords: primary hyperoxaluria; bone-marrow oxalosis; end-stage renal disease; nephrolithiasis; hemodialysis; cytopenia
Introduction
A buildup of abnormal substances in the blood and their deposition in different organs, including the bone marrow, can result from inborn metabolic disorders [1]. The uncommon genetic disorder known as primary hyperoxaluria (PH) affects glyoxylate metabolism, leading to excessive oxalate production and widespread calcium-oxalate deposition (oxalosis) [2]. Nephrocalcinosis and recurrent kidney stones are typically caused by changes in the urinary tract, particularly in the renal parenchyma [3]. Oxalate can build up all over the body and impact other tissues, including the bone marrow, as renal function declines [4]. Here, we describe the case of a 22-year-old man who had a history of nephrolithiasis, developed end-stage renal disease (ESRD), and was undergoing kidney transplant evaluation. Unexpectedly low blood cell counts occurred during his evaluation. and had a bone-marrow biopsy performed; the histological examination showed needle-shaped, birefringent crystals accompanied by a foreign-body giant-cell reaction and focal marrow fibrosis, which confirmed bone-marrow oxalosis and supported a diagnosis of primary hyperoxaluria [5]. This case underscores the need to consider systemic oxalosis in dialysis patients who have early-onset kidney stone disease and unexplained blood-related issues [5].
Case Report
A male patient, age 22, who had been suffering from kidney stones since he was a young child, was diagnosed with terminal kidney failure. He has been receiving maintenance hemodialysis twice a week for the past two years due to a progressive decline in his kidney function. In addition, he had undergone two kidney stone removal nephrolithotomy surgeries as a child. When routine pre-transplant tests revealed thrombocytopenia, the patient was evaluated for kidney transplantation. According to a complete blood count, the hemoglobin level was 11.4g/dL, the hematocrit was 41.5%, the mean corpuscular volume (MCV) was 97.9fL, the mean corpuscular hemoglobin (MCH) was 26.9pg, the total white blood cell count was 4.53×10⁹/L, the absolute neutrophil count was 3.35×10⁹/L, and the platelet count was 86×10⁹/L.
The inexplicable blood cell deficiency prompted a bone marrow aspiration and biopsy. There were many osteoblasts and osteoclasts close to bony trabeculae, along with localized trabecular resorption, in the bone marrow, which was found to be normocellular to slightly hypercellular. Additionally, there were areas of marrow fibrosis. Many needle-like, refractile crystals were grouped together in the marrow spaces, encircled by a massive cell reaction that is characteristic of a foreign body. These crystals exhibited strong birefringence when exposed to polarized light, which is consistent with the buildup of calcium oxalate (bone marrow oxalosis). The patient was diagnosed with Primary Hyperoxaluria with bone marrow oxalosis based on the patient's early-stage chronic kidney disease, long history of kidney stones, and unique bone marrow findings.
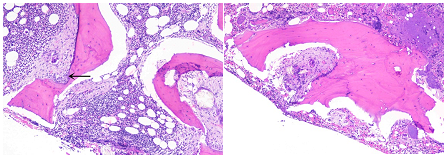
Figure 1: Bone trephine section shows hematopoietic areas surrounded by foreign body giant cells, fibrosis and also cartilage hypertrophy at ×40.
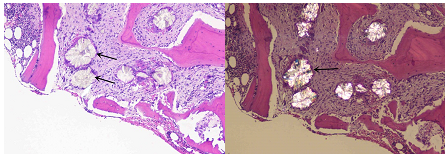
Figure 2: Bone marrow showing oxalate crystals can be seen in bone marrow with extensive starburst crystal deposition. Photographed in partly polarized light at ×40.
Discussion
Primary hyperoxaluria, or PH, is a genetic disorder that messes with the way the liver handles glyoxylate. That leads to way too much oxalate in the body, which ends up forming calcium-oxalate deposits—mainly in the kidneys [2]. Over time, if nothing stops it, people keep getting kidney stones and nephrocalcinosis, which slowly destroys kidney tissue and eventually causes end-stage renal disease. When the kidneys can’t filter properly anymore, oxalate builds up in the blood.(6) Then you get systemic oxalosis, where oxalate starts piling up all over—bones, blood vessels, heart, skin, and sometimes even the bone marrow [7].
Bone-marrow oxalosis doesn’t show up often, but it’s an important thing to watch for if someone with chronic kidney disease keeps having unexplained low blood counts and a history of kidney stones. When it hits the bone marrow, you see these sharp, needle-like crystals that really stand out under polarized light. The body reacts to them, usually with these big foreign-body giant cells and patches of fibrosis [8]. Over time, that messes with how the marrow works, sometimes leading to failure. People can end up with anemia, low platelets, low white cells, or even all three at once. You don’t see bone marrow biopsy reports for oxalosis very often, but when you do, it matters—especially if doctors can’t run certain blood tests or genetic studies. Sometimes, spotting it in the marrow is what really breaks the case [9].
From a diagnostic perspective, the case we discuss emphasizes a practical approach for clinicians: in a young individual with early-onset recurrent kidney stones who has progressed to chronic kidney disease/end-stage renal disease and presents with unexplained blood cell deficiencies, consider systemic oxalosis as a potential diagnosis and obtain targeted tissue samples (bone marrow or bone biopsy) if non-invasive tests do not provide clear answers. If feasible, biochemical assessments (urine oxalate, plasma oxalate) and genetic testing for AGXT/GRHPR/ HOGA1 mutations should be conducted to differentiate between PH1, PH2, and PH3 and inform treatment decisions; however, these tests might not be accessible or interpretable in patients on dialysis who are anuric, thereby highlighting the importance of histological examination [10].
When it comes to treatment, you’re really aiming for two things at once: lower the body’s oxalate levels and crystals, and deal with any organ problems that have popped up [11]. Dialysis can clear out some of the oxalate in the blood, but it’s not enough for PH1 because the liver just keeps making more [12]. For some people with certain AGXT gene variants, vitamin B6 (that’s pyridoxine) can make a big difference by cutting down how much oxalate the body produces, so it’s definitely worth a shot while sorting out a long-term plan [13].
Conclusion
In the end though, the only real cure for PH1 is a liver and kidney transplant—sometimes both at once, sometimes the liver first, then the kidney. This fixes the liver’s metabolic problem and brings kidney function back. The sooner this happens, before oxalate has really built up everywhere, the better the chances for a good outcome [14].
We’ve seen some clear limitations in our case and others—mostly, slow diagnoses and trouble getting genetic or enzyme tests. These problems pop up a lot in places with fewer resources, which really puts the spotlight on the value of histopathology. When we document cases diagnosed through bone marrow exams, we help other clinicians notice marrow oxalosis, add to what we know about its blood-related features, and maybe even speed up diagnosis for the next patient. Bottom line: marrow oxalosis doesn’t show up often, but when it does, it matters. Spotting it should push doctors to run a targeted metabolic workup right away and start making plans for real treatment. That’s how we can stop more permanent organ damage before it happens.
Declarations
Conflict of interest
None
Patient’s consent
Telephonic informed consent was obtained from the patient.
Competing interest
The authors declared no conflict interest.
Author’s contribution
Ms, sb, km: contributed to the design, drafting and critical revision of the manuscript.
All authors approved the final version of the manuscript to be published.
Disclaimer (artificial intelligence)
Author(s) hereby declares that no generative ai technologies such as large language models (chatgpt, copilot, etc.) And text-to-image generators have been used during the writing or editing of this manuscript.
References
- Agana M, Frueh J, Kamboj M, Patel DR, Kanungo S. (2018). Common metabolic disorder (inborn errors of metabolism) concerns in primary care practice. Ann Transl Med., 6(24):469.
Publisher | Google Scholor - Harambat J, Fargue S, Bacchetta J, Acquaviva C, Cochat P. (2011). Primary hyperoxaluria. Int J Nephrol. 2011:864580.
Publisher | Google Scholor - Habbig S, Beck BB, Hoppe B. (2011). Nephrocalcinosis and urolithiasis in children. Kidney International. 80(12):1278-1291.
Publisher | Google Scholor - Ermer T, Eckardt KU, Aronson PS, Knauf F. (2016). Oxalate, inflammasome, and progression of kidney disease. Curr Opin Nephrol Hypertens. 25(4):363-371.
Publisher | Google Scholor - Nematollahi P, Mohammadizadeh F. (2015). Primary Hyperoxaluria Diagnosed Based on Bone Marrow Biopsy in Pancytopenic Adult with End Stage Renal Disease. Case Reports in Hematology, 2015(1):402947.
Publisher | Google Scholor - Edvardsson VO, Goldfarb DS, Lieske JC, Beara-Lasic L, Anglani F, Milliner DS, et al. (2013). Hereditary causes of kidney stones and chronic kidney disease. Pediatr Nephrol., 28(10):1923-1942.
Publisher | Google Scholor - d'Izarny-Gargas T, Dang J, Grünenwald A, Mechref Z, Besson FL, Ferlicot S, et al. (2024). Bone Marrow Oxalosis. Kidney Int Rep. 9(4):1139-1141.
Publisher | Google Scholor - d'Izarny-Gargas T, Dang J, Grunenwald A, Mechref Z, Besson F, Ferlicot S, et al. (2024). Bone Marrow Oxalosis. Kidney International Reports. 9.
Publisher | Google Scholor - Sohail M, Rafique Y, Nawaz A, Sabir S. (2021). Primary Hyperoxaluria with Renal Failure and Bone Marrow Oxalosis in a young Patient in Pakistan. Pakistan Journal of Kidney Diseases, 1.
Publisher | Google Scholor - Mandrile G, Beck B, Acquaviva C, Rumsby G, Deesker L, Garrelfs S, et al. (2023). Genetic assessment in primary hyperoxaluria: why it matters. Pediatr Nephrol. 38(3):625-634.
Publisher | Google Scholor - Milliner DS, Eickholt JT, Bergstralh EJ, Wilson DM, Smith LH. (1994). Results of long-term treatment with orthophosphate and pyridoxine in patients with primary hyperoxaluria. N Engl J Med. 331(23):1553-1558.
Publisher | Google Scholor - Yamauchi T, Quillard M, Takahashi S, Man NK. (2024). Oxalate removal by daily dialysis in a patient with primary hyperoxaluria type 1. Nephrology Dialysis Transplantation, 16(12):2407-2411.
Publisher | Google Scholor - Dejban P, Lieske JC. (2022). New therapeutics for primary hyperoxaluria type 1. Curr Opin Nephrol Hypertens., 31(4):344-350.
Publisher | Google Scholor - Siegal D, Su WS, DaBreo D, Puglia M, Gregor L, Gangji AS. (2011). Liver-kidney transplantation in primary hyperoxaluria type-1: case report and literature review. Int J Organ Transplant Med., 2(3):126-132.
Publisher | Google Scholor





















