

Case Report
A Giant Extraosseous Ewing Sarcoma with Life-Threatening Airway Obstruction: A Case Report and Literature Review
- Berrios Mejia Juan Alberto *
- Contreras Rodriguez Felipe de Jesus
- Alcaraz Lopez Juan Gerardo
- Camacho Alamo Oscar Joseph
Pulmonology and Thoracic Surgery Department, Mexican Social Security Institute, Spetialty Hospital-Western Médical Center, Jalisco.
*Corresponding Author: Berrios Mejia Juan Alberto, Pulmonology and Thoracic Surgery Department, Mexican Social Security Institute, Spetialty Hospital-Western Médical Center, Jalisco.
Citation: Alberto B.M.J, Jesus C.R.F, Gerardo A.L.J, Joseph C.A.O. (2023). A Giant Extraosseous Ewing Sarcoma with Life-Threatening Airway Obstruction: A Case Report and Literature Review. Journal of Clinical Surgery and Surgical Research, BRS Publishers. 2(1); DOI: DOI: 10.59657/2992-9989.brs.23.007
Copyright: © 2023 Berrios Mejia Juan Alberto, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: March 14, 2023 | Accepted: March 30, 2023 | Published: April 05, 2023
Abstract
Extraosseous Ewing sarcoma is a malignant mesenchymal tumor. It can arise from any location; however, it generally appears in the paravertebral regions or in soft tissues of the extremities. It generally follows a rapid course and has high rates of recurrence. Pretreatment evaluation of the extension of disease is paramount since this is the most important factor in the outcome. Treatment consists of local surgery and neoadjuvant chemotherapy with some cases requiring radiotherapy. We describe the biggest tumor of EES primary to the lung parenchyma, with extensive tumor infiltration and the unique feature of airway obstruction. The tumor was successfully resected, and the patient treated with chemotherapy and radiotherapy. Extraosseous Ewing sarcoma of the lung is an extremely rare disease with less than 40 cases described in the literature.
Keywords: extraosseous ewing sarcoma; primitive neuroectodermal tumor; airway obstruction; pulmonary sarcoma
Introduction
Ewing sarcoma is a highly aggressive cancer, with a survival of 70-80% for patients with standard-risk and localized disease and ~30% for those with metastatic disease.
Case Report
A 24-year-old male, former vulcanizer and foundry worker, presented with a week history of thoracic pain with radiation to the scapula, fatigue, and dyspnea. Additionally, he reported nocturnal intermittent fevers. He denied other chronic diseases, allergies, or exposure to toxic substances. At the presentation he was tachycardic, tachypneic, and with slight hypotension. On examination, on the left hemithorax chest movements were diminished, had no breath sounds, and was dull to percussion.
Contrast chest computed tomography showed a total left lung collapse due to the presence of a giant mass occupying approximately 70% of the hemithorax, with dimensions of 178 x 145 x 85 mm on the longitudinal, anteroposterior y transverse planes respectively. The tumor had well-defined borders, was slightly heterogeneous, and had greater central contrast uptake in the venous phase. A pleural effusion occupying approximately 30 on the same side was also observed. A diaphragmatic decline of almost 90 mm is also noted. No extra thoracic or soft tissue extension was detected by this means.
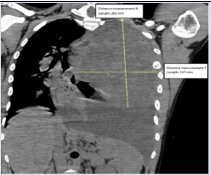
Figure 1: Thorax CT at presentation in sagittal view demonstrating height and width dimensions of the tumour.
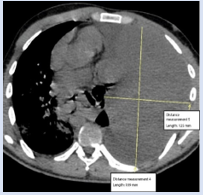
Figure 2: Thorax CT at presentation in axial view depicting tumour dimensions. An ipsilateral small pleural effusion is also noted.
A CT-guided biopsy of the mass was performed and atypical cells with round nucleus, finely dispersed chromatin, and scant cytoplasm were found. Surgery was performed through median sternotomy with extension to left anterior inframammary thoracotomy. Dissection of the antero-superior mediastinum is completed. Dissection of the adhesions of the chest wall is was complete with an ultrasonic energy device (HARMONIC ACE®+7 scissors, Johnson & Johnson Medical Devices). Once mass mobilization was achieved, Extensive lung infiltration of the tumor was found, and involvement of the left main pulmonary artery was encountered. Intrapericardial pneumonectomy was performed with ligation of the left pulmonary artery with a stapler (Endo GIA™ Reload with Tri-Staple™ Technology, Medtronic), 60mm Vascular cartridge, as well as both veins and the bronchus. In the Chest wall, a block resection of ribs [5,6,7] was completed, and the chest wall was reconstructed with a 15 x 10 cm polypropylene mesh. During surgery 700 mL of pleural fluid was also drained. Bleeding was estimated at 2000 mL. He was admitted to the UCI after surgery.
The specimen was 20 cm wide, 16 cm, and 15.5 cm, ovoid, multilobulated, and brown. It contained the sixth rib, lung, and tumor. The tumor was 17 cm in height, 15 cm in width, and 4 cm wide. Staininig for CD99 and FLI1 was positive in tumor cells.
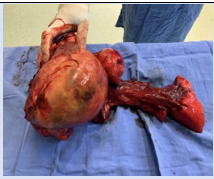
Figure 3: Pathological specimen containing tumor, lung, and the fifth to seventh rib.
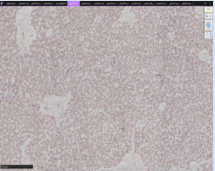
Figure 4: Stain for FLI1 demonstrates diffuse nuclear expression.
Biopsy showed a malignant neoplasm of round, small and hyperchromatic cells. The nucleus was round, with sub membranous chromatin, and punctiform nucleolus. Abundant necrosis with an irregular pattern was also identified with zones of hemorrhage. The diagnosis of primitive neuroendocrine tumor (extraosseous Ewing sarcoma). He was treated with adjuvant chemotherapy with cisplatin and etoposide, and radiotherapy with 45 Gy. Currently, at 18 months of follow-up, he is asymptomatic, with a functional class ECOG 2, and attends quarterly to the outpatient pulmonology, radio oncology, and medical oncology clinics.
Discussion
Extraosseous Ewing sarcoma (EES) is a malignant mesenchymal tumor. It is grouped under the undifferentiated small round cell sarcomas of bone and soft tissue category on the current WHO classification. 1 Ewing sarcoma (EES) represents ~6% to 47% of all Ewing tumors [2]. Genetic studies have suggested that EESs are in the same family as primitive neuroectodermal (PNET) tumors. These studies have demonstrated reciprocal translocation of t (11; 22) (q24; q12) in approximately 95% of patients, with the remainder demonstrating t (21; 22) (q22; q12) translocation [33]. In general terms, this tumor is more commonly found in older children/adults and often carries a poor prognosis and frequent recurrences [4,5,6]. Patients with EES often note a rapidly growing soft-tissue mass, with approximately one-third causing pain due to compression of adjacent structures. The most common primary sites of disease include the trunk (paravertebral regions) (32%), extremities (mostly in the lower extremities) (26%), head and neck (18%), retroperitoneum (16%), and other sites (9%) [7,8,9]. There is no evidence yet of any familial or environmental influence [3,4,7,9,10]. In the Surveillance, Epidemiology, and End Results (SEER) Program database, patients with extraosseous Ewing sarcoma were most commonly at the age range of 20-39 years, female, of non-White race, have more axial primary sites (and less pelvic primary site) involvement compared with patients with skeletal Ewing sarcoma [8].
Radiographic features
Radiographs may demonstrate a nonspecific soft-tissue mass in proximity to a bone without gross osseous involvement [7,9,10]. Computed tomography (CT) usually shows a soft tissue mass with similar attenuation as skeletal muscle. Magnetic resonance (MR) is the standard test used for the initial diagnosis and local staging and fluorodeoxyglucose-positron emission tomography (FDG-PET) is used for the detection of metastasis, to assess response to chemotherapy, and to detect of recurrent disease [11,13].
A definitive diagnosis is made through histopathological evaluation of a CT-guided or ultrasound-guided core-needle biopsy [14].
Pathological characteristics
EES often appears gray-yellow or gray-tan on gross pathological specimens, with lobulations and a soft texture. Histopathology confirms EES via monotonous proliferation of small round blue cells solidly packed with intracellular glycogen which may have indent nuclei with a lightly colored eosinophilic cytoplasm [7]. Cystic/necrotic regions demonstrating rich vascularity, and areas of hemorrhage are often present. Membrane staining is almost always positive for CD99. [15,16]. Histologic staining for Friend Leukemia Integration 1 (FLI1) protooncogene (more than 10% of tumor cells), demonstrating the translocation [11,22]. will also provide a definitive diagnosis. [17,15].
Treatment and prognosis
At present management is largely based on extrapolation and assumption of similar behavior with Ewing sarcoma [18]. Treatment often involves neoadjuvant chemotherapy, local control with surgery and/or radiotherapy [5,10,19-24]. Chemotherapy includes cyclic combinations, incorporating vincristine, doxorubicin, cyclophosphamide, alternating with iphosphamide, and etoposide and occasionally actinomycin D [18,24]. Increased tumor burden and distant metastasis at presentation are associated with poor overall survival [5]. The 5-year survival for patients with localized disease is around 60% and 40% for patients with metastatic disease [24].
Conclusion
Extraosseous Ewing sarcoma is an extremely rare disease. The main site for primary disease remains the chest with more than half of the cases. The diagnostic approach is similar to other tumors of the chest; however, early diagnosis is vital to establish optimal treatment due to the overall poor prognosis and frequent recurrences. This entity should be included in the differential diagnosis of a mass in the chest or abdomen. Treatment is well established. Treatment consists of local surgery and/or radiotherapy treatment followed by systemic chemotherapy.
Acknowledgments
Every author was responsible for the gathering of information, writing, and publication of the case report.
The authors would like to thank the patient and his family for their support during the entire care process.
The patient signed informed consent for management and publication of his medical information.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest concerning the research, authorship, and/or publication.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
- Kallen ME, Hornick JL. (2021). The 2020 WHO Classification: What’s New in Soft Tissue Tumor Pathology? Am J Surg Pathol. 45(1):1-23.
Publisher | Google Scholor - Fizazi K, Dohollou N, Blay JY, et al. (1998). Ewing’s family of tumors in adults: multivariate analysis of survival and long-term results of multimodality therapy in 182 patients. J Clin Oncol. 16(12):3736-3743.
Publisher | Google Scholor - Nagaraj P, H SC, Rao R, Manohar S. Extra skeletal Soft Tissue Ewing’s Sarcoma with Variant Translocation of Chromosome t (4; 22) (q35; q12)-A Case Report. J Orthop Case Rep. 3(4):12-15.
Publisher | Google Scholor - el Weshi A, Allam A, Ajarim D, et al. (2010). Extraskeletal Ewing’s sarcoma family of tumours in adults: analysis of 57 patients from a single institution. Clin Oncol (R Coll Radiol). 22(5):374--381.
Publisher | Google Scholor - Muratori F, Mondanelli N, Pelagatti L, et al. Clinical features, prognostic factors and outcome in a series of 29 extra-skeletal Ewing Sarcoma. Adequate margins and surgery-radiotherapy association improve overall survival. J Orthop. 21:236-239.
Publisher | Google Scholor - Raney RB, Asmar L, Newton WA, et al. (1997). Ewing’s sarcoma of soft tissues in childhood: a report from the Intergroup Rhabdomyosarcoma Study, 1972 to 1991. Journal of Clinical Oncology. 15(2):574-582.
Publisher | Google Scholor - Murphey MD, Senchak LT, Mambalam PK, Logie CI, Klassen-Fischer MK, Kransdorf MJ. (2013). From the radiologic pathology archives: ewing sarcoma family of tumors: radiologic-pathologic correlation. Radiographics. 33(3):803-831.
Publisher | Google Scholor - Applebaum MA, Worch J, Matthay KK, et al. (2011). Clinical features and outcomes in patients with extraskeletal Ewing sarcoma. Cancer. 117(13):3027-3032.
Publisher | Google Scholor - O’Keeffe F, Lorigan JG, Wallace S. (1990). Radiological features of extraskeletal Ewing sarcoma. Br J Radiol. 63(750):456-460.
Publisher | Google Scholor - Marjara J, Hilli J, Davis RM, Bhat AP. (2020). Metastatic retro-crural lymph nodes from transitional cell carcinoma of bladder successfully treated with single session cryoablation. Radiol Case Rep. 15(8):1197-1201.
Publisher | Google Scholor - Meyer JS, Nadel HR, Marina N, et al. (2008). Imaging guidelines for children with Ewing sarcoma and osteosarcoma: a report from the Children’s Oncology Group Bone Tumor Committee. Pediatr Blood Cancer. 51(2):163-170.
Publisher | Google Scholor - Franzius C, Sciuk J, Daldrup-Link HE, Jürgens H, Schober O. (2000). FDG-PET for detection of osseous metastases from malignant primary bone tumours: comparison with bone scintigraphy. Eur J Nucl Med. 27(9):1305-1311.
Publisher | Google Scholor - Völker T, Denecke T, Steffen I, et al. (2007). Positron emission tomography for staging of pediatric sarcoma patients: results of a prospective multicenter trial. J Clin Oncol. 25(34):5435-5441.
Publisher | Google Scholor - Brinkhuis M, Wijnaendts LC, van der Linden JC, et al. (1995). Peripheral primitive neuroectodermal tumour and extra-osseous Ewing’s sarcoma; a histological, immunohistochemical and DNA flow cytometric study. Virchows Arch. 425(6):611-616.
Publisher | Google Scholor - Sandberg AA, Bridge JA. (2000). Updates on cytogenetics and molecular genetics of bone and soft tissue tumors: Ewing sarcoma and peripheral primitive neuroectodermal tumors. Cancer Genet Cytogenet. 123(1):1-26.
Publisher | Google Scholor - Fellinger EJ, Garin-Chesa P, Glasser DB, Huvos AG, Rettig WJ. (1992). Comparison of cell surface antigen HBA71 (p30/32MIC2), neuron-specific enolase, and vimentin in the immunohistochemical analysis of Ewing’s sarcoma of bone. Am J Surg Pathol. 16(8):746-755.
Publisher | Google Scholor - Folpe AL, Chand EM, Goldblum JR, Weiss SW. (2001). Expression of Fli-1, a Nuclear Transcription Factor, Distinguishes Vascular Neoplasms from Potential Mimics. The American Journal of Surgical Pathology. 25(8):1061-1066.
Publisher | Google Scholor - J. Sybil Biermann MAHMPCWCM et al. NCCN Clinical Practice Guidelines in Oncology - Bone Cancer. Springer International Publishing.
Publisher | Google Scholor - el Weshi A, Allam A, Ajarim D, et al. (2010). Extraskeletal Ewing’s sarcoma family of tumours in adults: analysis of 57 patients from a single institution. Clin Oncol (R Coll Radiol), 22(5):374-381.
Publisher | Google Scholor - Galyfos G, Karantzikos GA, Kavouras N, Sianou A, Palogos K, Filis K. (2016). Extraosseous Ewing Sarcoma: Diagnosis, Prognosis and Optimal Management. Indian J Surg. 78(1):49-53.
Publisher | Google Scholor - El-Essawy MT. (2009). Extraskeletal Ewing’s sarcoma. Saudi Med J. 2009;30(6):840-843.
Publisher | Google Scholor - Zagar TM, Triche TJ, Kinsella TJ. (2008). Extraosseous Ewing’s sarcoma: 25 years later. J Clin Oncol. 26(26):4230-4232.
Publisher | Google Scholor - Senne J, Davis R, Yasin J, Brimmo O, Evenski A, Bhat AP. Computed tomography guided radio-frequency ablation of osteoid osteomas in atypical locations. Indian J Radiol Imaging. 29(3):253-257.
Publisher | Google Scholor - Rodriguez-Galindo C, Spunt SL, Pappo AS. (2003). Treatment of Ewing sarcoma family of tumors: current status and outlook for the future. Med Pediatr Oncol. 40(5):276-287.
Publisher | Google Scholor - Bernstein M, Kovar H, Paulussen M, et al. (2006). Ewing’s sarcoma family of tumors: current management. Oncologist. 11(5):503-519.
Publisher | Google Scholor














