

Research Article
A Comprehensive Review of Mitochondrial Genetic Mutations in Melas Syndrome
- Shahin Asadi *
- Amir Shokri
- Mohadeseh Zoughi
Medical Genetics Director of the Division of Medical Genetics and Molecular Pathology Research, Center of Complex Disease, United States.
*Corresponding Author: Shahin Asadi,Medical Genetics Director of the Division of Medical Genetics and Molecular Pathology Research, Center of Complex Disease, United States.
Citation: Asadi S, Shokri A, Zoughi M. (2024). A Comprehensive Review of Mitochondrial Genetic Mutations in Melas Syndrome. International Journal of Medical Case Reports and Reviews, BioRes Scientia Publishers. 3(2):1-7. DOI: 10.59657/2837-8172.brs.24.046
Copyright: © 2024 Shahin Asadi, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: March 27, 2024 | Accepted: March 08, 2024 | Published: March 13, 2024
Abstract
MELAS syndrome (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes) is a rare disorder that begins in childhood, usually between the ages of two and fifteen, and mostly involves the nervous system and muscles. Symptoms of MELAS syndrome usually begins between the ages of two and fifteen years, but cases with delayed onset have also been reported between fifteen and forty years and late onset cases after forty years. In approximately 75% of cases, the onset of this disorder is before the age of 20. People with MELAS syndrome has lactic acid accumulation in the blood (lactic acidosis), which can lead to vomiting, abdominal pain, fatigue, muscle weakness, and difficulty breathing. This accumulation of lactic acid has also been observed in the spinal fluid and in the brain. MELAS is caused by mutations in mitochondrial DNA (mtDNA).
Keywords: melas syndrome; mtdna; metabolic disorder; lactic acid; polg1 gen
Introduction
Overview of MELAS Syndrome
MELAS syndrome (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes) is a rare disorder that begins in childhood, usually between the ages of two and fifteen, and mostly involves the nervous system and muscles. The most common initial symptoms of seizures are frequent headaches, loss of appetite and frequent vomiting. Episodes of stroke with temporary muscle weakness on one side of the body (hemiparesis) can also occur, and this can lead to altered consciousness, decreased vision and hearing, loss of motor skills, and mental retardation. MELAS is caused by a mutation in mitochondrial DNA, and in one patient, this syndrome is associated with a mutation in the nuclear gene POLG1 [1].
Clinical Signs and Symptoms of MELAS Syndrome
Symptoms of MELAS syndrome usually begins between the ages of two and fifteen years, but cases with delayed onset have also been reported between fifteen and forty years and late onset cases after forty years. In approximately 75% of cases, the onset of this disorder is before the age of 20. Symptoms and physical findings related to MELAS syndrome are very different between affected people in the same family and between different families. A distinctive feature of MELAS syndrome is the recurrence of stroke-like episodes. It is now thought that a lack of a compound called nitric oxide in the small blood vessels of the brain may be responsible for strokes. Short stature and hearing loss may be present, and fatigue and difficulty in tolerating exercise may be early symptoms [2].
People with MELAS syndrome has lactic acid accumulation in the blood (lactic acidosis), which can lead to vomiting, abdominal pain, fatigue, muscle weakness, and difficulty breathing. This accumulation of lactic acid has also been observed in the spinal fluid and in the brain. In some cases, affected people will experience a slow decline in mental function (dementia) or a reduced ability to communicate with speech, writing, or signs (aphasia). People with MELAS syndrome may have periods of confusion and hallucinations, often due to a previous fever (febrile illness) or headache. Less common symptoms include involuntary muscle spasms (myoclonus), impaired muscle coordination (ataxia), cardiomyopathy, diabetes mellitus, depression, bipolar disorder, digestive problems, and kidney problems [1].
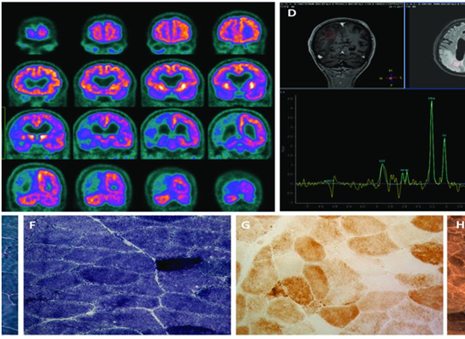
Figure 1: Radiological and microscopic images of disorders associated with MELAS syndrome.
Etiology of MELAS Syndrome
MELAS is caused by mutations in mitochondrial DNA (mtDNA). Mutations affecting mtDNA genes are inherited from the mother. mtDNA, which is found in sperm cells, is usually lost during fertilization, and as a result, all human mtDNA comes from the mother. The affected mother transmits this mutation to all her children, but only her daughters transmit this mutation to their children. Mitochondria, found by the hundreds or thousands in the body's cells, especially in muscle and nerve tissue, carry the blueprints for regulating energy production [1,2].
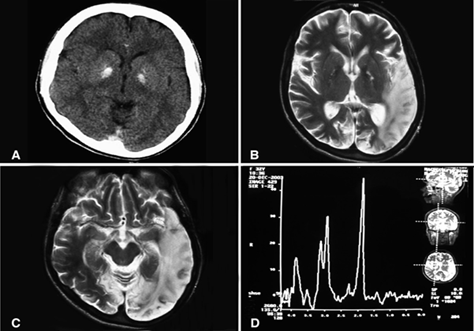
Figure 2: Radiological images of brain disorder in MELAS syndrome.
Both normal and mutated mtDNA can exist in the same cell, a condition known as heteroplasmy. The number of defective mitochondria may be more than the number of normal mitochondria. Symptoms may not appear in any given generation until the mutation affects a significant portion of the mtDNA. Unequal distribution of natural and mutated mtDNA in different tissues can affect different organs of the same family members. This can lead to various symptoms in affected family members [1,2]. Mutations in the mtDNA MT-TL1 gene are associated with MELAS in approximately 80% of cases. Mutations in MT-TQ, MT-TH, MT-TK, MT-TS1, MT-ND1, MT-ND5, MT-ND6 and MT-TS2 are also associated with MELAS syndrome. Some cases of MELAS syndrome appear to occur as a result of a new spontaneous mutation in a mitochondrial gene and are not inherited. In addition, a mutation in a nuclear gene (POLG1) located in the long arm of chromosome 15, 15q26.1, was associated with MELAS syndrome in one case [1,2].
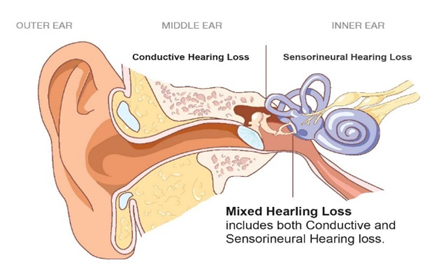
Figure 3: Schematic of the anatomy of the inner ear, middle ear and outer ear.
Frequency of MELAS Syndrome
MELAS syndrome is a rare disorder that affects men and women equally. Although rare, MELAS syndrome is probably the most common type of mitochondrial myopathy caused by mutations in mtDNA. Some researchers believe that mitochondrial myopathies may be underdiagnosed and underdiagnosed in the general population, making it difficult to determine the true frequency of disorders such as MELAS syndrome [1,2].
Disorders Associated with MELAS Syndrome
Kearns-Sayre syndrome is a rare neuromuscular disorder characterized by three main findings: progressive paralysis of some eye muscles (chronic progressive external ophthalmoplegia), abnormal accumulation of pigment (pigmentation) on the nerve-rich membrane covering the eye (retinitis). unusual pigmentosa) which leads to chronic inflammation, progressive degeneration and wear and tear of some eye structures (retinal pigmentary degeneration) and heart diseases such as heart block. Other findings may include muscle weakness, short stature, hearing loss, or loss of the ability to coordinate voluntary movements (ataxia) due to problems affecting a part of the brain (cerebellum). In some cases, Kearns-Sayre syndrome may be associated with other disorders or conditions [1,3].
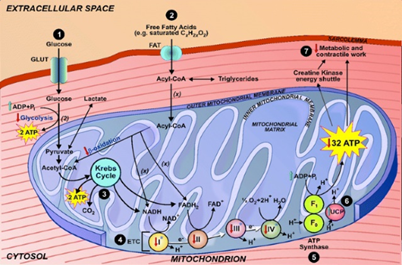
Figure 4: Schematic of the molecular and biochemical mechanism of the mitochondrial organelle.
MERRF syndrome (myoclonus epilepsy with red fibers) is a very rare disorder that begins in childhood and affects the nervous system and muscles as well as other body systems. The distinguishing feature of MERRF syndrome is sudden, brief, jerking spasms that can affect the arms and legs or the whole body (myoclonic convulsions). In addition, people with MERRF syndrome may have muscle weakness (myopathy), impaired ability to coordinate movements (ataxia), seizures, and a slow decline in mental function (dementia). Short stature, optic nerve degeneration (optic atrophy), hearing loss and cardiomyopathy are also common symptoms. Abnormal muscle cells are present and appear as irregular red fibers when stained and viewed microscopically. MERRF is caused by mutations in mitochondrial DNA [1,3].
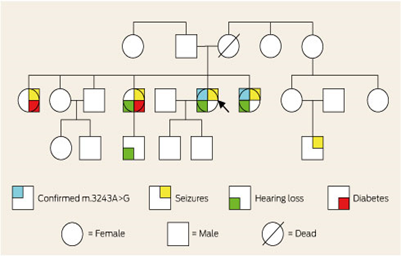
Figure 5: Schematic of the genetic genealogy sample of MELAS syndrome sufferers along with related disorders.
Leigh disease is a rare genetic neurometabolic disorder. It is characterized by degeneration of the central nervous system (eg, brain, spinal cord, and optic nerve). The symptoms of Leigh's disease usually start between three months and two years of age. Symptoms are associated with gradual neurological deterioration and may include loss of previous motor skills, loss of appetite, vomiting, irritability, or seizure activity. As Leigh disease progresses, symptoms may include general weakness, lack of muscle tone (hypotonia), and episodes of lactic acidosis, which may lead to impaired respiratory and renal function [1,3]. It seems that there are several different types of genetically determined enzyme defects that can cause Leigh disease. Most people with Leigh disease have defects in mitochondrial energy production, such as deficiency of mitochondrial respiratory chain enzyme or pyruvate dehydrogenase complex. In most cases, Leigh disease is inherited as an autosomal recessive trait. However, recessive inheritance and X-linked mitochondria have also been noted [1,3].
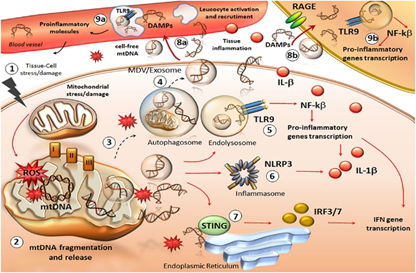
Figure 6: Schematic of the stages of energy production by the mitochondrial organelle.
Diagnosis of MELAS Syndrome
MELAS is diagnosed based on clinical findings and molecular genetic tests. Clinical examination may include measurement of lactate and pyruvate concentration and CSF protein, which are increased in MELAS syndrome. Brain imaging techniques such as magnetic resonance imaging (MRI) may be used to look for stroke-like lesions and magnetic resonance spectroscopy (MRS) to look for lactate peaks in the brain. An electrocardiogram may be used to diagnose heart rhythm disorders and an echocardiogram may be used to diagnose cardiomyopathy. Muscle biopsy usually shows red fibers [1,4]. MELAS-associated mtDNA mutations can usually be detected in white blood cells, but because of heteroplasmy (causes), other tissue samples such as skin, hair follicles, urinary sediment, and skeletal muscle may be required. Compared to blood, skin and hair follicles, urine sediment has the best performance for mutation detection [1,4].
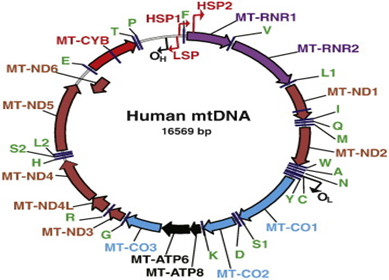
Figure 7: Schematic of the physical map of human mtDNA.
MELAS Syndrome Treatment Pathways
There is no specific treatment available for MELAS syndrome. Anticonvulsant drugs are used to help prevent and control seizures associated with MELAS syndrome. Valproic acid should not be used as an anticonvulsant. Cochlear implantation has been used to treat sensorineural hearing loss. Sometimes therapeutic methods are used to increase energy production by mitochondria and reduce the effects of this disease. Coenzyme Q10 and L-carnitine have been useful in some patients. In patients with mitochondrial myopathy in general, moderate treadmill exercise may improve aerobic capacity and decrease resting lactate levels. The use of intravenous l-arginine has been reported to improve symptoms during acute episodes of stroke. It has been reported that the use of oral arginine reduces the recurrence of stroke-like episodes in the asymptomatic period. Genetic counseling is recommended for affected people and their families [1,5].
Research Treatments
Medicines such as carnitine, coenzyme Q10, menadione, ascorbic acid, riboflavin, tamin, nicotinamide, creatine monohydrate, idebenone, succinate and dichlorostat have been useful in individual patients, but more studies are needed to prove their effectiveness. Arginine and citrulline are being investigated as potential treatments to reduce brain damage caused by stroke [1,5].
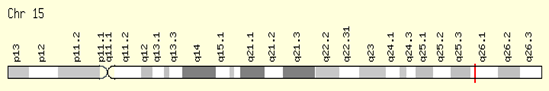
Figure 8: A schematic of the physical map of chromosome number 15, where the POLG1 gene is located in the long arm of this chromosome as 15q26.1.
Columbia University in New York City is seeking study participants for a double-blind, placebo-controlled clinical trial of idebenone in MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes). A phase IIa study compares two different doses of an experimental drug, idebenone, administered over a one-month period to determine the drug's effectiveness. People with MELAS and the 3243 mutations, between the ages of 8 and 65, may be eligible. The primary goal of the clinical trial is to determine whether idebenone has an effect on brain lactate as measured by magnetic resonance spectroscopy (MRS). MRS is performed in an MRI scanner and is safe and usually well tolerated. An additional objective is to study the safety and tolerability of idebenone in subjects with MELAS [1,5]
Baylor College of Medicine and Texas Children's Hospital are recruiting people with MELAS syndrome for two clinical studies. The first study aims to measure nitric oxide in affected people and check whether giving arginine or citrulline increases the formation of nitric oxide or not. Nitric oxide is thought to be useful in the recovery and prevention of stroke, therefore, if arginine or citrulline are shown to increase the formation of nitric oxide, they can be used to prevent and improve stroke in patients with used to MELAS syndrome. Diabetes is common in MELAS syndrome, and the purpose of the second study is to evaluate how sugar is broken down and made in the body of affected people. This may lead to a better understanding of the causes of diabetes in MELAS, which can affect the prognosis and treatment of diabetes in people with MELAS. Adults and children with MELAS syndrome and carriers of m.3243 A>G mutation can participate. Study participants will be admitted to the General Clinical Research Center (GCRC) at Texas Children's Hospital, where nitric oxide and glucose production will be measured by injection of stable isotopes, a safe method [1,5].
Discussion
The most common initial symptoms of seizures are frequent headaches, loss of appetite and frequent vomiting. Episodes of stroke with temporary muscle weakness on one side of the body (hemiparesis) can also occur, and this can lead to altered consciousness, decreased vision and hearing, loss of motor skills, and mental retardation. People with MELAS syndrome has lactic acid accumulation in the blood (lactic acidosis), which can lead to vomiting, abdominal pain, fatigue, muscle weakness, and difficulty breathing. This accumulation of lactic acid has also been observed in the spinal fluid and in the brain. In some cases, affected people will experience a slow decline in mental function (dementia) or a reduced ability to communicate with speech, writing, or signs (aphasia). People with MELAS syndrome may have periods of confusion and hallucinations, often due to a previous fever (febrile illness) or headache. Mutations affecting mtDNA genes are inherited from the mother. mtDNA, which is found in sperm cells, is usually lost during fertilization, and as a result, all human mtDNA comes from the mother. The affected mother transmits this mutation to all her children, but only her daughters transmit this mutation to their children. Mutations in the mtDNA MT-TL1 gene are associated with MELAS in approximately 80% of cases. Mutations in MT-TQ, MT-TH, MT-TK, MT-TS1, MT-ND1, MT-ND5, MT-ND6 and MT-TS2 are also associated with MELAS syndrome. There is no specific treatment available for MELAS syndrome.
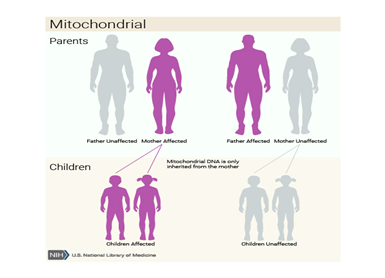
Figure 9: Schematic of the mitochondrial (maternal or cytoplasmic) inheritance pattern that MELAS syndrome follows.
References
- Asadi S. (2024). Pathology in Mitochondrial Genetic Book, Amidi Publications, Iran.
Publisher | Google Scholor - Deschauer M, Tennant S, Rokicka A, et al. (2007). MELAS associated with mutations in the POLG1 gene. Neurology, 68(20):1741-1742.
Publisher | Google Scholor - Scaglia F, Northrop JL. (2006). The mitochondrial myopathy encephalopathy, lactic acidosis with stroke-like episodes (MELAS) syndrome: a review of treatment options. CNS Drugs, 20(6):443-64.
Publisher | Google Scholor - Koga Y, Akita Y, Nishioka J, et al. (2005). L-arginine improves the symptoms of strokelike episodes in MELAS. Neurology, 64(4):710-712.
Publisher | Google Scholor - Scaglia F. (MELAS Syndrome. eMedicine Journal.
Publisher | Google Scholor














