

Case Report
A Case Report of The Adult Asymptomatic Alcapa Syndrome
1Cardiology Fellow, Tabba Heart Institute, Karachi, Pakistan.
2Consultant Cardiologist, Tabba Heart Institute, Karachi, Pakistan.
*Corresponding Author: Ubaidullah Popal,Cardiology Fellow, Tabba Heart Institute, Karachi, Pakistan.
Citation: Marium M, Ubaidullah P, Adeelurehman S. (2024). A Case Report of The Adult Asymptomatic Alcapa Syndrome, International Journal of Cardiology Research and Reports, BRS Publishers. 2(1); DOI: 10.59657/2996-3109.brs.24.004
Copyright: © 2024 Ubaidullah Popal, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: July 18, 2023 | Accepted: December 29, 2023 | Published: January 16, 2024
Abstract
Coronary artery anomalies encompass a broad spectrum of anatomical and pathophysiological abnormalities. Although the rare, anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA) syndrome is a well-known disease. This rare congenital syndrome primarily is characterized by the anomalous origin of the left coronary artery from the pulmonary artery. In this report, we present a 49 years old man with no known comorbid admitted due to acute gastroenteritis. In the course of diagnosis, initially, ECG showed poor R wave progression. CT coronary angiogram was performed to access coronary arteries which revealed the anomalous origin of the left coronary artery system from the main pulmonary artery.
Keywords: ALCAPA; anomalous left coronary artery; coronary angiography
Introduction
Coronary artery anomalies span a varied spectrum of anatomical and pathophysiological abnormalities. Most of these anomalies have no hemodynamic or pathophysiological significance and therefore, majority of these cases are detected accidentally. However, significant heart anomalies that as a consequence have shunt volume, myocardial ischemia or ventricular tachyarrhythmia’s which can lead to sudden cardiac death, emphasize the importance of early detection. Early detection of rare cardiovascular anomalies and taking early-stage surgery are crucial in preventing undesirable outcomes [1]. Anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA), although rare, but is a well-known disease [2]. This rare congenital syndrome primarily is characterized by the anomalous insertion of the left coronary artery into the pulmonary artery [3]. Most of these cases can be seen in the pediatric population, especially in the first year, presenting with the symptoms of heart failure and sudden cardiac death due to chronic myocardial ischemia [3]. However, there are pieces of evidence that a certain number of patients with this rare syndrome survive to adulthood [2]. The characteristics of adult ALCAPA syndrome are a compensatory formation of collaterals between the right coronary artery (RCA) that in physiological conditions has the origin from the aorta and left coronary artery (LCA). ALCAPA syndrome was traditionally diagnosed at angiography. This diagnostic modality enables direct visualization of the coronary arteries. The development of computed tomographic (CT) angiography and magnetic resonance imaging (MRI) enabled precise and non-invasive diagnosis. CT angiography provides direct visualization of the left coronary artery arising from the main pulmonary artery. As the MRI has the ability to assess myocardial viability, MRI takes a very important role in the treatment as a prognostic factor for the necessity of surgical repair [4].
Case Presentation
49 years old man with no known comorbidities was admitted due to acute gastroenteritis. ECG was done as a routine workup and showed low voltage R wave progression, due to which the patient was referred to a cardiologist for further diagnostic evaluation. The patient denied history of palpitations, chest pain, and shortness of breath. A Dobutamine stress test was conducted revealing multivessel ischemia despite the patient being asymptomatic, the ejection fraction (EF) was reported to be between 40-45%.
To further evaluate the myocardial ischemia, angiography was recommended. The right coronary artery was found to be dilated, branching into multiple inter-coronary collaterals of various sizes (figure1 and 2) communicating with the left coronary system. However, attempts to cannulate the left coronary artery were unsuccessful. (Figure 3). Aortogram showed RCA arising from right coronary cusp, but left coronary artery was not found to be arising from the left coronary cusp. CT coronary angiogram was done to access coronary arteries which showed the anomalous origin of the left coronary artery system from the main pulmonary artery (figure 5-8). Moreover, MPI was done to evaluate the degree of ischemia but no signs of ischemia were found. Therefore, the patient was kept on medical treatment with regular follow-ups.
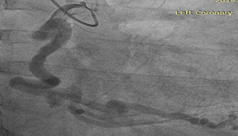
Figure 1
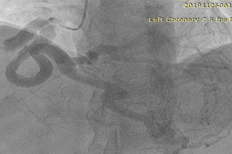
Figure 2
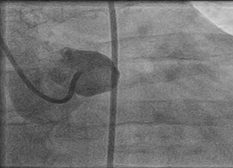
Figure 3
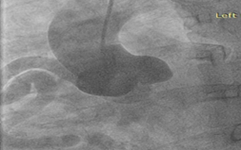
Figure 4
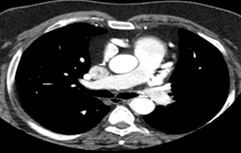
Figure 5
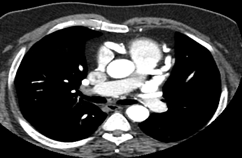
Figure 6
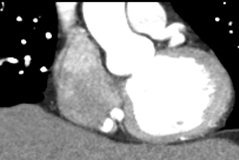
Figure 7
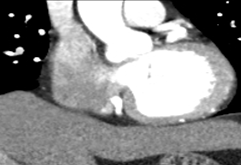
Figure 8
Discussion
The Anomalous origin of the left coronary artery from the pulmonary artery syndrome also known as Bland-White-Garland syndrome is extremely rare congenital cardiovascular anomalies that affect 1 in 300,000 cases of live births [4,5]. This syndrome accounts for around 0.5% of all congenital heart anomalies [4]. A considerable number of cases are isolated, while around 5% of cases might be associated with other congenital heart anomalies [4]. Based on above, it can be concluded that the incidence of the asymptomatic presentation of ALCAPA syndrome in adulthood, as in the case presented, is exceptionally rare at 0.26% [6].
The main feature of ALCAPA syndrome is the coronary steal phenomenon, in which a left-to-right shunt causes abnormal perfusion of the left ventricle of the heart [4]. The most common clinical presentation is dyspnea, angina pectoris, dilated cardiomyopathy, myocardial ischemia, heart failure, and sudden death in adulthood or death in childhood [6]. In case ALCAPA syndrome is left unmanaged, it carries a mortality of 90% with in the first year [4].
After coronary arteries have been developed, maturing and extension occur on the heart’s surface. In the physiological circumstances, the right coronary artery (RCA) originates from the anterior side of the coronary sinus and extends to the right atrioventricular groove. The left main coronary artery bifurcates after 1cm of its origin from left coronary sinus into the left anterior descending artery and left circumflex coronary artery [7]. The most common origin of the anomalous coronary arteries is a contralateral side, while the less common is from noncoronary sinus or pulmonary artery. Due to this “crossing” of coronary arteries, these anomalous blood vessels are longer, and usually without branching [7]. The absence of ALCAPA signs and symptoms at birth is due to negligible difference between pulmonary blood pressure and pressure in the systemic circulation. As pulmonary pressure decrease after a few months, symptoms and signs can be detected. The degree of collateral circulation between two coronary arteries determines the degree of myocardial ischemia. Therefore, the patients who developed rich collateral circulation are classified as “adult” type of ALCAPA syndrome as in this case, while the patients with no or poor collaterals are classified as the “infantile” type. Pulmonary hypertension develops gradually in those with the adult type of this syndrome. The pathophysiological mechanism of pulmonary hypertension in ALCAPA syndrome is that the blood passes retrogradely due to left-to-right shunt, from the right to the left coronary artery through collateral blood vessels, finishing at the left main coronary artery and then into the pulmonary artery [6]. Given this specific case, as the patient was asymptomatic throughout life, and developed collateral vessels capable of providing oxygen to the myocardium even during physical activity, it is understandable why the discovery was completely accidental. [8]. ALCAPA syndrome was traditionally diagnosed by coronary angiography. On angiography, dilated, tortuous right coronary arteries can be observed accompanied by collateral filling of the left coronary artery. Feature in ECG which can suggest ALCAPA syndrome is left axis deviation, hypertrophy of left ventricle, poor R wave progression, and abnormal Q waves in leads I and aVL. On echocardiography, a dilated right coronary artery with an uninterrupted flow of blood from the left coronary artery to the pulmonary artery can be detected. Nowadays, modern diagnostic methods such as cardiac MRI and CT angiography are frequently used for detecting cardiac abnormalities as high-resolution imaging provides direct visualization of the left coronary artery arising from the pulmonary artery ALCAPA syndrome is standardly managed by surgery [4]. The main goal of surgery is to reconstruct a two-coronary system providing physiological blood flow without shunting [4,6]. In the case of the asymptomatic adults who developed rich network of collaterals, survival without surgical repair is possible. Therefore, this patient was started on guideline directed medical therapy (GDMT), with regular follow-ups, as this will provide regular surveillance and surgery could be performed if the deterioration of the patient's condition eventually occurs. [8]. The patient has been followed for the last 4 years, is asymptomatic and living a healthy life.
Conclusion
Our case pertains to a 49-year-old asymptomatic patient whose congenital heart anomaly, ALCAPA syndrome, was serendipitously diagnosed following an unrelated gastroenteritis episode. This case is of considerable significance due to the patient's age and the extraordinary fact that they remained asymptomatic throughout their entire life. The significance of our case lies in showcasing the potential of ALCAPA syndrome to remain undiagnosed due to the development of rich collateral circulations. Hence, we underscore the necessity for maintaining a high degree of suspicion, even when faced with unrelated medical presentations, to timely diagnose and manage such life-threatening anomalies.
References
- Heermann P, Heindel W, Schuelke C. (2017). Coronary Artery Anomalies: Diagnosis and Classification based on Cardiac CT and MRI (CMR)-from ALCAPA to Anomalies of Termination. RoFo: Fortschritte auf dem Gebiete der Rontgenstrahlen und der Nuklearmedizin, 189(1):29-38.
Publisher | Google Scholor - Boutsikou M, Shore D, Li W, Rubens M, Pijuan A, Gatzoulis MA, Babu-Narayan SV. (2018). Anomalous left coronary artery from the pulmonary artery (ALCAPA) diagnosed in adulthood: Varied clinical presentation, therapeutic approach and outcome. International journal of cardiology, 261:49-53.
Publisher | Google Scholor - Quah JX, Hofmeyr L, Haqqani H, Clarke A, Rahman A, Pohlner P, Radford D, Nicolae M. (2014). The management of the older adult patient with anomalous left coronary artery from the pulmonary artery syndrome: a presentation of two cases and review of the literature. Congenital heart disease, 9(6):185-194.
Publisher | Google Scholor - Peña E, Nguyen ET, Merchant N, Dennie C. ALCAPA syndrome: not just a pediatric disease. Radiographics, 29(2):553-565.
Publisher | Google Scholor - Dodge-Khatami A, Mavroudis C, Backer CL. (2002). Anomalous origin of the left coronary artery from the pulmonary artery: collective review of surgical therapy. The Annals of thoracic surgery, 74(3):946-955.
Publisher | Google Scholor - Zacharias M, Chandok D, Tighe D. (2015). A late presentation of an anomalous left coronary artery originating from the pulmonary artery (ALCAPA): a case study and review of the literature. Journal of cardiology cases, 11(2):56-59.
Publisher | Google Scholor - Danias PG, Stuber M, McConnell MV, Manning WJ. The diagnosis of congenital coronary anomalies with magnetic resonance imaging. Coronary artery disease. 2001 Dec 1;12(8):621-626.
Publisher | Google Scholor - Gouda P, Gouda J, Butler C, Welsh RC. Late presentation of an anomalous left coronary artery from the pulmonary artery treated with conservative surgical management with long-term cardiac magnetic resonance imaging follow-up. SAGE open medical case reports, 5.
Publisher | Google Scholor












