

Case Report
A Case of Familial Takayasu Arteritis
- Narra Lavanya 1*
- Immaneni Sathyamurthy 2
- Kanthallu Narayanamoorthy Srinivasan 3
- Sasikala Subramanian 4
1Interventional Cardiologist, General Medicine, Dr NB Cardiology Apollo main hospital, Tamil Nādu, India.
2Senior Consultant in Interventional Cardiologist, Apollo main hospital, Tamil Nādu, India.
3Senior Consultant Interventional Cardiologist, General Medicine, Dr NB Cardiology Apollo Hospital, Tamil Nādu, India.
*Corresponding Author: Narra Lavanya, Interventional Cardiologist, General Medicine, Dr NB Cardiology Apollo main hospital, Tamil Nādu, India.
Citation: Lavanya N, Sathyamurthy I, Kanthallu N. Srinivasan, Subramanian S. (2024). A Case of Familial Takayasu Arteritis, Journal of Clinical Cardiology and Cardiology Research, BioRes Scientia Publishers. 3(1); DOI: 10.59657/2837-4673.brs.24.023
Copyright: © 2024 Narra Lavanya, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: February 01, 2024 | Accepted: February 21, 2024 | Published: March 06, 2024
Abstract
Takayasu’s Arteritis (TA) is a chronic granulomatous large vessel vasculitis. It commonly involves aorta and its main branches. Although TA is seen in both sexes, it is more prevalent in females and common among Asians. This ethnic preponderance in Asia suggests a possible role of genetic factors in the aetiology of TA. Familial clustering of TA although rare has been reported. In this report, we describe familial TA in a mother and daughter presenting with varying manifestations and who were managed appropriately.
Keywords: takayasu’s arteritis; vasculitis; familial clustering; aortoatreritis
Introduction
Takayasu’s Arteritis (TA) is an idiopathic large vessel vasculitis predominantly affecting elastic arteries such as the aorta, its major branches and also pulmonary arteries [1]. TA affects women predominantly in the 2nd to 3rd decade of their life and has been frequently reported from Japan, East Asia, India, and Mexico. The aetiology of TA is thought to be multifactorial. Infections, genetic factors and autoimmunity may play a role in its pathogenesis. The genetic contribution to the pathogenesis of TA is mainly supported by the association with a specific pattern of human leukocyte antigen (HLA) class I and II and with non-HLA genes involved in immune response [2]. The susceptibility to TA is contributed by HLA-B52 and TNF-α-308 A/G polymorphisms [3].
Case report
We report TA in a mother and daughter. A 26-year-old lady was referred to our center with history of angina and dyspnea on exertion New York Heart Association (NYHA class III) and syncope of 3 months duration (two episodes; not related to exertion). Clinical examination revealed absent bilateral radial pulses and feeble (1+) bilateral ulnar and brachial artery pulses [4]. Blood pressure could not be recorded in the upper limbs and lower limb blood pressure was 130/40mmHg. She had bilateral carotid and abdominal bruits and a bruit over the back.
C-reactive protein (CRP) titers were elevated to 50mg/L (normal Less-than5 mg/L). Her ECG revealed sinus rhythm with T wave inversions in antero-lateral precordial leads. Chest X-ray was normal. Echocardiogram showed regional wall motion abnormality (RWMA) involving mid apical septum, left ventricular (LV) apex and inferior wall with grade I Mitral valve prolapse (MVP) and LV ejection fraction (EF) of 55%. Her Coronary angiogram showed ostial left main coronary artery (LMCA) total occlusion. Left anterior descending (LAD), Left circumflex (LCX) were seen filling retrogradely via collaterals. The right coronary artery (RCA) was dominant and normal. CT aortogram showed evidence of diffuse wall thickening involving aortic root, ascending aorta, arch and descending aorta (Figure 1). Left common carotid artery showed 50 % stenosis, left subclavian artery showed total occlusion and the right subclavian artery showed 80 % discrete lesion. Both renal arteries showed non obstructive plaques at ostia. All the features were confirmatory of TA.
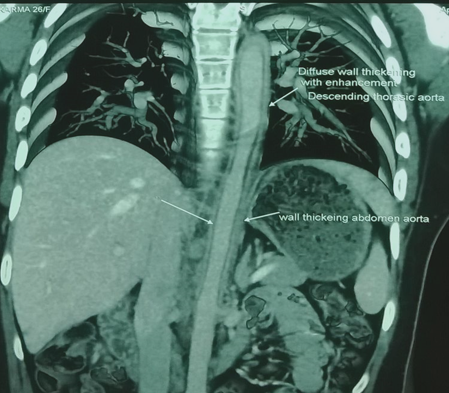
Figure 1: CT aortogram showed diffuse wall thickening involving aortic root, ascending aorta, arch and descending aorta.
In view of her symptoms, it was decided by the heart team to go ahead with off-pump coronary artery bypass grafting (CABG). Intra-operatively there was diffuse thickening of aortic wall with plaques near the root and origin of the innominate artery. The left internal mammary artery (IMA) was non pulsatile. The right IMA was 1.25 mm and thickened. A saphenous vein graft (SVG) was anastomosed to LAD and proximal anastomosis was done to right ventricular (RV) branch of RCA (as the RV branch was close to origin of RCA). Post-operative recovery was uneventful. The patient was started on steroids and methotrexate and was discharged in a stable state.
Patient delivered a baby via caesarean section 2 years after CABG which was uneventful. 640 slice CT coronary angiogram in 2021 revealed patent venous graft between RV branch of RCA and LAD (Figure 2a, b). The most recent follow up was in 2023. Patient was asymptomatic and clinically stable. Lab investigations revealed normal erythrocyte sedimentation rate (ESR) and CRP (Less-than2.86mg/L). Echocardiogram showed grade I MVP with normal LV function. The patient was continued on steroids and methotrexate as a part of maintenance immunosuppression. This case was previously reported by us [5].
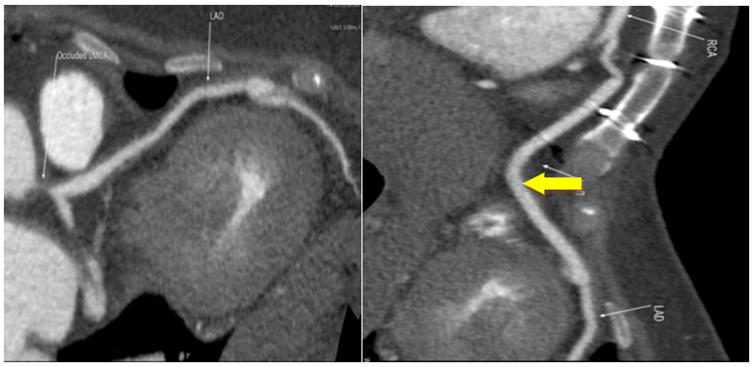
Figure 2: a. The LM was occluded and LAD and diagonal branches, the LCx and OM branches revealed no significant stenosis. b. The venous graft between RV branch of RCA and LAD was patent (yellow arrow).
Her mother is a 49-year-old lady, a known hypertensive and presented with history of dyspnoea, chest discomfort and palpitations on exertion (NYHA) II since 1996. Clinical examination revealed bilateral absent ulnar pulsations with normal blood pressure in both arms. She had bilateral carotid bruit, abdominal bruit and an early diastolic murmur in the aortic area. Her lab investigations revealed elevated ESR and Creatine phosphokinase (CPK). Her Electrocardiogram (ECG) showed Atrial fibrillation (AF) with controlled ventricular rate and her echocardiogram showed normal ventricular function with mild to moderate aortic regurgitation (AR) and Mitral regurgitation (MR). CT aortogram revealed mild diffuse wall thickening with dilatation involving the ascending aorta and aortic arch. There was diffuse narrowing of the descending thoracic aorta with small areas of sacculation. Celiac artery ostium showed an 80% stenosis. The diagnosis of TA was confirmed.
She was treated with beta-blockers and was started on anticoagulation for AF. She has been on regular follow up since then. During the most recent follow-up in 2023, patient was asymptomatic and clinically stable. Lab investigations revealed mildly elevated ESR and normal CPK. CT aortogram revealed diffuse narrowing of descending thoracic aorta with multiple outpouchings and wall irregularities. There was mild narrowing of left subclavian and carotid artery and focal narrowing at the origin of celiac axis. (Figure 3a, b) Patient was continued on beta-blockers and anticoagulation. Immunosuppression was not initiated due to stable disease status.
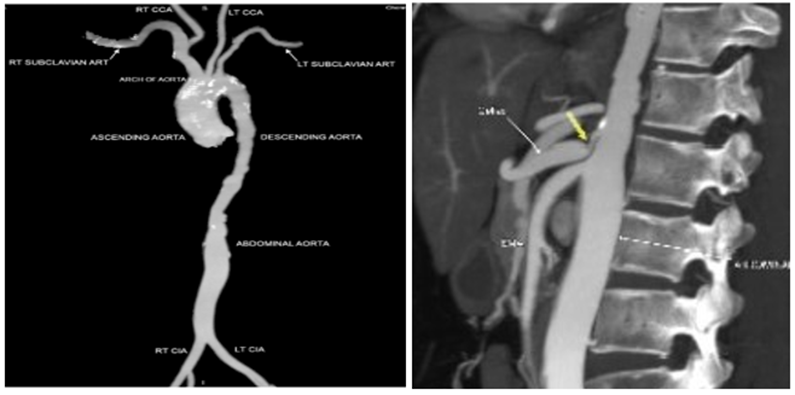
Figure 3: a. CT aortogram revealed diffuse narrowing of descending thoracic aorta with multiple outpouchings and wall irregularities. b. yellow arrow showing the focal 80% narrowing of the celiac axis origin.
Discussion
Familial TA is a relatively rare condition with isolated case reports. Japan reported the greatest number of familial TA cases with sisters being the most commonly affected siblings. The most common genetic association was found with HLA B-52 [6]. Identification of candidate genes in TA can be attempted using homozygosity mapping [7]. In our case report, the daughter was referred for her class III symptoms. She had absent limb pulses with elevated inflammatory markers. She was evaluated with CT aortogram and found to have diffuse aortic thickening and CT-CAG showed ostial LMCA total occlusion. She underwent CABG and is doing well on continued medical management. Her mother on the other hand though symptomatic since a longer time was not diagnosed until recently as she was not evaluated thoroughly. She was found to have reduced limb pulses, elevated inflammatory markers and diffuse aortic thickening on CT aortogram. She was continued on medical management and is asymptomatic on follow-up.
Most of the familial cases revealed that many of the affected individuals were only diagnosed later in life and subtle clinical signs were missed early [8]. In our case, the daughter was diagnosed at a younger age due to significant symptoms, but her mother was diagnosed only later in life when detailed examination was done. The presentation of the two differed wherein the daughter underwent CABG and the mother did well on medical management. The differential diagnosis of TA includes giant cell arteritis (in the elderly), fibromuscular dysplasia (in the young) and vasculitis. The age of onset is important in coming to an appropriate diagnosis. In our case the similarity of clinical signs in the mother and daughter point in the direction of TA even though the mother was diagnosed at a much later age. The limitation of our case report is that genetic analysis could not be performed.
Conclusion
Our case report demonstrates the importance of maintaining a high index of suspicion while evaluating family members of affected patient. Adequate clinical evaluation using four-limb blood pressure measurements and pulse examination should be made mandatory in such cases. The mother could have been diagnosed and treated earlier had the presence of reduced limb pulses and bruit were identified sooner. The occurrence of TA in the mother and daughter demonstrates the possibility of a genetic etiology. Since it is a rare association, we are reporting these cases.
References
- Lupi-Herrera E, Sánchez-Torres G, Marcushamer J, Mispireta J, Horwitz S, et al. (1977). Takayasu’s arteritis. Clinical study of 107 cases. American Heart Journal. 93(1):94–103.
Publisher | Google Scholor - Renauer P, Sawalha AH. (2017). The genetics of Takayasu arteritis. Presse Med. 46(7-8 Pt 2):e179–187.
Publisher | Google Scholor - Chen S, Luan H, Li L, Zeng X, Wang T, et al. (2017). Relationship of HLA-B*51 and HLA-B*52 alleles and TNF-α-308A/G polymorphism with susceptibility to Takayasu arteritis: a meta-analysis. Clin Rheumatol. 36(1):173–181.
Publisher | Google Scholor - Hill RD, Smith RB. (1990). Examination of the Extremities: Pulses, Bruits, and Phlebitis. In: Walker HK, Hall WD, Hurst JW, editors. Clinical Methods: The History, Physical, and Laboratory Examinations [Internet]. 3rd ed. Boston: Butterworths.
Publisher | Google Scholor - Sathyamurthy I, Lavanya N, Srinivasan KN, Girinath MR. (2022). Coronary-to-coronary bypass grafting in aortoarteritis: a case report. Indian J Thorac Cardiovasc Surg. 38(5):511–514.
Publisher | Google Scholor - Morishita KA, Rosendahl K, Brogan PA. (2011). Familial Takayasu arteritis - a pediatric case and a review of the literature. Pediatric Rheumatology. 9(1):6.
Publisher | Google Scholor - Nalls MA, Guerreiro RJ, Simon-Sanchez J, Bras JT, Traynor BJ, et al. (2009). Extended tracts of homozygosity identify novel candidate genes associated with late-onset Alzheimer’s disease. Neurogenetics. 10(3):183–190.
Publisher | Google Scholor - Deniz A, Yildiz F, Aktas H, Berk GI, Erken E, et al. (2013). Familial Takayasu arteritis in a mother and daughter: a report of two cases. Herz. 38(1):93–96.
Publisher | Google Scholor











