

Case Report
Rare Extranodal Manifestations of Rosai-Dorfman Disease: Single Organ Involvement as a form of Onset
1Rheumatology, Hospital Central de Reconquista, Santa Fe, Argentina.
2Rheumatology Service JM Cullen Hospital, Santa Fe, Argentina.
*Corresponding Author: Gallo Jessica Romina, Rheumatology, Hospital Central de Reconquista, Santa Fe, Argentina.
Citation: Gallo J. Romina, Sergio P. (2024). Rare Extranodal Manifestations of Rosai-Dorfman Disease: Single Organ Involvement as a form of Onset. Journal of Clinical Rheumatology and Arthritis, BioRes Scientia Publishers. 2(1):1-6. DOI: 10.59657/2993-6977.brs.24.013
Copyright: © 2024 Gallo Jessica Romina, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: March 04, 2024 | Accepted: March 19, 2024 | Published: March 27, 2024
Abstract
Rosai-Dorfman disease is a rare non-Langerhans cell histiocytosis that mainly affects bilateral cervical lymphadenopathy, with the head and neck being the most affected regions. 43% of patients manifest extranodal disease. This report presents 3 cases of an uncommon form of onset of this disease, such as pulmonary and skin involvement. It is very important to take into account the differential diagnoses of these presentations, which can undoubtedly increase the delay in diagnosis and the initiation of early treatment.
Keywords: rosai-dorfman disease; sinus histiocytosis; extranodal manifestations; rosai-dorfman disease of the lung; cutaneous rosai-dorfman disease
Introduction
Rosai-Dorfman Disease (RDD), also known as sinus histiocytosis, is a rare condition characterized by heterogeneous presentations, with lymphadenopathy as its cardinal sign. While the disease typically affects lymph nodes, extranodal involvement has been observed in approximately 43% of cases, with 23% presenting exclusively with extranodal manifestations. Aside from concurrent lymph node involvement, the most common extranodal sites, in descending order, include the skin, soft tissues, nasal cavity and sinuses, eyes, orbits and ocular adnexa, bone, salivary gland, central nervous system, kidney, intrathoracic structures, and cardiac structures, among others [1].
Cutaneous lesions are found in 10% of cases of systemic RDD, but cutaneous involvement as an isolated manifestation is observed in 3% of cases, similar to intrathoracic involvement [1,2,3]. Systemic RDD shows a predilection for males and young individuals, especially those of African descent, while the diagnosis of cutaneous RDD is typically specific to middle-aged women of Asian or Caucasian origin. Diagnosis is based on histologic findings, which include histiocytic infiltration with emperipolesis and positive immunohistochemistry for S-100 protein, lysozyme, a1-antitrypsin, CD68 and CD163 with negative Langerhans cell markers (CD1a and langerin). Histology may vary at each stage of evolution [4]. We present three cases with rare extranodal manifestations, cutaneous and intrathoracic, as a form of onset and without involvement of another organ.
Case 1
A 19-year-old female patient was referred due to skin lesions with a 1.5-year evolution, initially treated as allergic dermatitis. The patient presented with peri-oral skin lesions and some on the cheeks, characterized by papules and pustules, some exhibiting acneiform features, along with a photosensitive malar rash. Physical examination revealed the mentioned skin lesions (Figure 1 ab). No signs of hair loss, oral or mucosal ulcers, arthritis, malar rash, or adenopathies were observed. Laboratory results showed normal erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), rheumatoid factor (RF), antinuclear antibodies (ANA), Anti-dsDNA antibodies, antibodies to extractable nuclear antigens (ENA), negative results, normal complement, IgE, IgG, IgA, IgM, and IgG subclasses 1, 2, 3, 3, and 4. Viral serology was negative. The skin biopsy reported a granulomatous inflammatory infiltrate composed of histiocytes and multinucleated giant cells. Immunohistochemistry results were positive for CD68, S100, and CD163, negative for CD1a, and non-reactive for IgG4. Final diagnosis: Rosai-Dorfman disease with cutaneous onset. Treatment was initiated with Prednisone 10 mg per day, methotrexate 15 mg, and folic acid, resulting in a favorable response (figure 1-c).
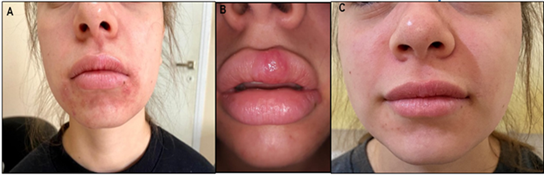
Figure 1: Cutaneous manifestation of Rosai-Dorfman. (A, B) Lesions Pre-treatment. (C) Lesions post-treatment.
Case 2
A 66-year-old male with a history of smoking presented with a medical history that included a presumptive diagnosis of cutaneous systemic lupus erythematosus (SLE) and Raynaud's phenomenon at 36 years of age. Physical examination revealed no lymphadenopathy, malar rash, skin lesions, hair loss, arthritis, oral or nasal ulcers, or Raynaud's phenomenon. Respiratory and abdominal examinations were unremarkable. Laboratory tests indicated anemia, leukopenia on two occasions (2100/3100), platelets, eosinophils, and normal renal function. Erythrocyte sedimentation rate (ESR) was at 66 mm/1 hour, and CRP was 8 mg/l (0-5 mg/l), both elevated. Rheumatoid factor, antineutrophil cytoplasmic antibody (ANCA), ANA, Anti-dsDNA antibodies, anti-Ro, anti-La, anti-Sm, anti-RNP, anticardiolipin, lupus anticoagulant, and B2 glycoprotein were negative, while C3 and C4 were within normal ranges. Serum IgE levels were elevated at 19309 mg/ dl (nv: 0-100 UI/ml), and IgA was 716 mg/dl (nv:70-400 mg/dl).
Immunoglobulin M, IgG, IgG subclasses (IgG1, IgG2, IgG3, IgG4), angiotensin-converting enzyme, and a normal proteinogram. Kappa and lambda chains by immunofixation, and Bence-Jones protein were negative. 18F-fluorodeoxyglucose (FDG)-PET/CT was conducted (Figure 2 a,b), revealing thickening of the interlobular septa and areas of hypoperfusion predominantly in the apices of the lungs. A dense, irregular image associated with converging traces towards the pleura was observed. Additionally, there was a ground-glass image, bronchiectasis, and pretracheal adenopathies, while the rest of the structures appeared normal. The echocardiogram was within normal limits. A pulmonary nodule and pleura biopsy were performed, revealing chronic plasmacytic inflammatory infiltrate, macrophages with hemosiderin, and dense, non-storiform fibrosis. Immunohistochemistry showed CD1a negativity, CD68 positivity, S100 positivity, IgG positivity, and IgG4: 5 cells per high-power field (HPF). Final diagnosis: pulmonary Rosai-Dorfman. Treatment was initiated with meprednisone 1 mg/kg associated with methotrexate 15 mg/week and folic acid. The patient was lost to follow-up 12 months after initiation.
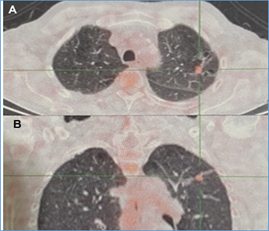
Figure 2:18FDG-PET CT-scan. (A, B) Showing radiotracer uptake in pretracheal adenopathies, pulmonary nodule, and pleura before treatment.
Case 3
A 47-year-old woman with a medical history of Gastric Stromal Tumor (GIST) diagnosed in 2018 and undergoing treatment with Imatinib for nine months. She presented acutely dry cough, progressive dyspnea, and thrombocytopenia. An 18F-fluorodeoxyglucose (FDG)-PET/CT scan (Figure 3 A, B) was performed, revealing multiple hypermetabolic ganglionic structures in the mediastinal region, segmental bronchi, subpleural regions, and bilateral jugulo-carotid region. Subpleural nodules were also detected in the right upper and lower lobes, along with small micronodules in the minor fissure and splenosis. A lymph node biopsy was conducted, which revealed granulomas with epithelioid histiocytes and multinucleated giant cells, accompanied by lymphocytic crowns but no necrosis. Preliminary diagnosis: Sarcoidosis, and the patient was referred to rheumatology. Physical examination revealed no palpable lymphadenopathy, hair loss, arthritis, oral or nasal ulcers, skin changes, or Raynaud's phenomenon. Respiratory examination indicated decreased bilateral air entry. Upon re-evaluation of the biopsy, immunohistochemistry was performed, showing CD1a negativity and positivity for S100, CD163, and CD68. Final diagnosis: Rosai-Dorfman disease. Treatment was initiated with meprednisone at 1 g/kg per day, with descending doses, in conjunction with methotrexate at 15 mg weekly and folic acid supplementation. After 10 months of treatment, an 18F-fluorodeoxyglucose (FDG)-PET/CT scan was requested, revealing an improvement in the imaging findings. However, some hypermetabolic lymphadenopathies persisted with a decreased SUV, although indicating ongoing activity. As a result, a dose of rituximab 1000 mg/m2/dose, 2 doses 2 weeks apart and then 6 months later was initiated. One year later, a subsequent 18F-fluorodeoxyglucose (FDG)-PET/CT scan demonstrated total improvement of the clinical picture.
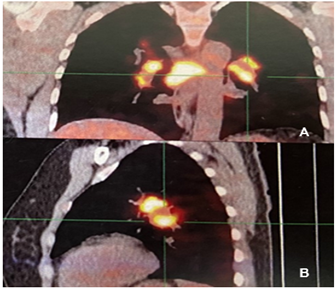
Figure 3: 18F-fluorodeoxyglucose (FDG)-PET/CT. (A, B) The PET/CT images before treatment displayed hypermetabolic lymph nodes and subpleural nodules
Discussion
The most recognized manifestation of RDD is the nodal form. In classic (nodal) RDD, most patients present with painless, massive, bilateral cervical lymphadenitis, often accompanied by intermittent fever, night sweats, and weight loss [5]. Leukocytosis, neutrophilia, elevated ESR, and polyclonal hypogammaglobulinemia may be observed, and they may resolve spontaneously or with treatment [5-7]. Histopathologically, nodal RDD is characterized by sinus expansion of large histiocytes with a large hypochromatic nucleus and prominent nucleolus. Emperopolesis, while a useful finding, is not necessary for diagnosis and may be focal, particularly in extranodal sites. It can also be observed focally in other histiocytoses such as Erdheim-Chester disease, juvenile xanthogranuloma, and malignant histiocytosis. On immunohistochemistry, nodal RDD typically shows positivity for S100, CD68, and CD163, and negativity for CD1a, in contrast to Langerhans cell histiocytosis (LCH) [5].
Extranodal RDD occurs in 43% of cases, sometimes without associated lymphadenopathy, which may or may not develop later in the course of the disease. In 23% of cases, extranodal involvement may be the sole presentation. This latter form of presentation was observed in our three patients [1,5,8]. Case 1 presented cutaneous lesions in the perioral region, lips, and cheeks, exhibiting features such as papules, pustules, small perioral vesicles, and painful small vesicles on the lips. Additionally, there were other lesions with acneiform characteristics on the cheeks. These manifestations are part of the broad spectrum of cutaneous manifestations. Skin involvement is heterogeneous, possibly presenting as macules, papules, nodules, or infiltrated erythematous or violaceous plaques, xanthomatous or yellowish-brown, isolated or disseminated. Lesions resembling slow-growing tumors, painless and non-pruritic, with colors ranging from yellow to red to brown, have also been described, along with lesions mimicking vasculitis or panniculitis. Pustular acneiform lesions, a manifestation also observed in our patient, were present as well [9,10]. The head and face are the most commonly affected regions in patients of all ages, followed by the trunk and extremities. Kong et al. divided the disease into three types: papulonodular type (79.5%), indurated plaque type (12.8%), and tumor type (7.7%). The lesion may develop gradually from the papulonodular type to the plaque type and finally to the tumor type [11,12].
The skin is involved in 10% of extranodal RDD cases and isolated cutaneous disease is rare (3%) [5]. This involvement affects a higher average age (43.5 years) than nodal RDD and is more frequent in female patients [13-15]. Due to the pleomorphism of the skin lesions, the complexity of the disease course, misleading histological patterns, and the absence of lymphadenopathy, diagnosis is often overlooked. Such situations occurred in our patient, resulting in a delay of almost one and a half years before the correct diagnosis was made [12]. As a result, the differential diagnosis encompasses a broad spectrum of pathologies, including granuloma annulare (GA), granulomatous diseases, acne vulgaris, rosacea, varicella-zoster virus infection, sarcoidosis, deep fungal infections, other cutaneous histiocytosis such as LCH, generalized eruptive histiocytoma, xanthogranuloma, reticulohistiocytosis, pyogenic granuloma, dermatofibroma, vasculitis, cutaneous lymphoma, and metastasis, among others [16,17]. Intrathoracic involvement, similar to isolated cutaneous involvement, has been described as infrequent, affecting only 2% of the respiratory tract, typically in young patients with an average age of 14 years [2,3,8,18].
Cartin-Ceba et al. described a series of 21 patients in which intrathoracic manifestations were present in 9 out of 21 patients (43%) with RDD. In 5 of these cases, intrathoracic involvement was confirmed based on the presence of mediastinal lymphadenopathy that could not be attributed to other etiologies. Confirmatory biopsies were obtained from other sites, predominantly cervical lymph nodes and skin enlargements. The most common intrathoracic manifestation of RDD was mediastinal lymphadenopathy in 6 patients (66%). Additionally, four patients (44%) exhibited respiratory tract involvement, such as cystic lung involvement, airway disease (including air trapping and bronchiectasis), nodules, pleural effusion with mediastinal adenopathy, and interstitial pulmonary infiltrates. No statistically significant differences were observed between the groups of patients with intrathoracic versus non-intrathoracic involvement in terms of age, time to diagnosis, sex, smoking history, mortality, and survival after diagnosis. Respiratory symptoms included dyspnea in 8 patients and cough in 5. The prognosis of these patients with intrathoracic manifestations was relatively favorable [3].
Chronic dry cough, progressive dyspnea or acute respiratory failure have also been described [2,5,18]. Our two patients with intrathoracic involvement presented with pulmonary and subpleural nodules, thickened septa, ground-glass opacities, bronchiectasis, and hilar and mediastinal lymphadenopathy. Only Case 2 was asymptomatic, while Case 3 exhibited symptoms such as dry cough and progressive dyspnea. Similar to cutaneous involvement, this presentation can mimic several pathologies, leading to differential diagnoses including Langerhans cell histiocytosis, tumors, lymphoma, sarcoidosis, IgG4-related disease with interstitial involvement, Erdheim-Chester disease, interstitial lung disease, or organizing pneumonia, as well as granulomatosis with polyangiitis, pulmonary disease related to rheumatoid arthritis, and mycobacterial and fungal infections. In contrast to nodal RDD, extranodal lesions histologically exhibit more fibrosis, fewer histiocytes, and less emperipolesis, thereby making diagnosis more challenging [5]. None of our patients presented emperopolesis, but foamy histiocytes, plasma cells, and abundant non-storiform fibrosis were observed [9]. Studies have shown the presence of IgG4+ and IgG4/IgG plasma cells in RDD cases, which may indicate a relationship with IgG4-related sclerosing diseases [19]. The immunohistochemistry of our patients showed no high-field IgG4 cells in one patient, but two of them had fewer than 10 high-field IgG4 cells. All of them presented positive staining for CD68, S100, and CD163, and negative staining for CD1a, confirming the diagnosis.
In general terms, RDD has a good prognosis, although its course is unpredictable and can range from spontaneous remission without intervention to persistent disease with episodes of remission and exacerbation. However, RDD affecting the lower respiratory tract can exhibit an aggressive phenotype, with a mortality rate of nearly 45%, and approximately 33% of cases show persistent and/or progressive disease [16,20]. Approximately 50% of patients do not require treatment, and spontaneous remission has been observed in approximately 20% of cases. Some benefit has been reported from the use of glucocorticoids and methotrexate alone or in combination with other agents, including systemic corticosteroids, 6-mercaptopurine inhibitors, imatinib, TNF-α inhibitors, IL-6 inhibitors, and Rituximab [21]. All our patients were treated with daily steroids at 1 mg/kg associated with weekly methotrexate at 15 mg. Cases 1 and 2 have remained in remission without treatment, and no symptomatic or radiological changes have been observed during a period of one year and a half. Only case 3 required the use of Rituximab due to persistent lymphadenopathy, with subsequent improvement.
Conclusion
Cutaneous and pulmonary involvement of RDD are rare manifestations of the condition, posing diagnostic challenges for both clinicians and pathologists due to their low frequency and heterogeneous clinical and histologic presentations [21]. Therefore, it is crucial to consider this disease, along with its rare organ involvement and extranodal presentation without lymphadenopathy, as a potential cause of symptoms. Laboratory, imaging, and histologic/immunohistochemical studies should be conducted to exclude other extracutaneous or extrapulmonary manifestations of the disease, as well as to rule out other pathologies. Early and accurate diagnosis of RDD is essential to initiate timely treatment and improve the prognosis of affected patients.
Declarations
Conflict of interest
The authors declare no conflict of interest
Informed Consent
Written informed consent was obtained from the patients who participated in this study.
No specific funding was received from any bodies in the public, commercial or not-for-profit sectors to carry out the work described in this article.
Acknowledgments
The authors are grateful to Mrs. Agostina Braidot for her help in translating the manuscript
References
- Shi SS, Sun YT, Guo L. (2009). Rosai-Dorfman disease of the lung: a case report and review of the literature. Chinese Medical Journal, 122(7):873-874.
Publisher | Google Scholor - Ohori NP, Yu J, Landreneau RJ, Thaete L, Kane K. (2003). Rosai-Dorfman disease of the pleura: a rare extranodal presentation. Hum Pathol, 34(11):1210-1211.
Publisher | Google Scholor - Cartin-Ceba R, Golbin JM, Yi ES, Prakash UB, Vassallo R. (2010). Intrathoracic manifestations of Rosai-Dorfman disease. Respir Med, 104(9):1344-1349.
Publisher | Google Scholor - Gawdzik A, Ziarkiewicz-Wro´blewska B, Chlebicka I, Jankowska-Konsur A. et al. (2021). Cutaneous Rosai-Dorfman Disease: A Treatment Challenge. Dermatol Ther (Heidelb), 11:1443-1448.
Publisher | Google Scholor - Abla O, Jacobsen E, Picarsic J, Krenova Z et al. (2018). Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood, 131(26):2877-2890
Publisher | Google Scholor - McClain KL, Bigenwald C, Collin M, et al. (2021). Histiocytic disorders. Nat Rev Dis Primers, 7(1):73.
Publisher | Google Scholor - Bruce-Brand C, Schneider JW, Schubert P. (2020). Rosai-Dorfman disease: an overview. J Clin Pathol, 73(11):697-705.
Publisher | Google Scholor - Foucar E, Rosai J, Dorfman R. (1990). Sinus histiocytosis with massive lymphadenopathy Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol, 7(1):19-73.
Publisher | Google Scholor - Quispetira J, Moisés C, Paredes A, Sánchez G, Pacheco M. et.al. (2009). Enfermedad de Rosai-Dorfman cutaneous: reporte de caso y revision de la literatura. Folia dermatol. Peru, 20(2):91-94.
Publisher | Google Scholor - Cabrera HN, Venegas MSF, Verdejo G, Casas JG. (2010). Enfermedad de Rosai-Dorfman: forma clínica acneiforme. Dermatol Argent, 16:23-26.
Publisher | Google Scholor - Kong YY, Kong JC, Shi DR, et al. (2007). Cutaneous Rosai–Dorfman disease: a clinical and histopathologic study of 25 cases in China. Am J Surg Pathol, 31(3):341-350.
Publisher | Google Scholor - Pan Zhang P, Liu F, Cha Y, Zhang X, Cao M. (2021). Self-Limited Primary Cutaneous Rosai-Dorfman Disease: A Case Report and Literature Review. Clinical, Cosmetic and Investigational Dermatology, 14:1879-1884.
Publisher | Google Scholor - FANG S, CHEN AJ. (2015). Facial cutaneous Rosai-Dorfman disease: A case report and literature review. Exp Ther Med, 9(4):1389-1392.
Publisher | Google Scholor - Thawerani H, Sanchez RL, Rosai J, Dorfman RF. (1978). The cutaneous manifestations of sinus histiocytosis with massive lymphadenopathy. Arch Dermatol, 114:191-197.
Publisher | Google Scholor - Ahmed A, Crowson N, Magro CM. (2019). A comprehensive assessment of cutaneous Rosai–Dorfman disease. Ann Diagn Pathol, 40:166-173.
Publisher | Google Scholor - Lu CI, Kuo TT, Wong WR, Hong HS. Clinical and histopathologic spectrum of cutaneous Rosai-Dorfman disease in Taiwan. J Am Acad Dermatol, 51(6):931-939.
Publisher | Google Scholor - Wang KH, Chen WY, Liu HN, Huang CC, Lee WR. (2006). Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol, 154:277-286.
Publisher | Google Scholor - Umairi R AL, Blunt D, Hana W, Cheung M, Anastasia Oikonomou A. (2018). Rosai-Dorfman Disease: Rare Pulmonary Involvement Mimicking Pulmonary Langerhans Cell Histiocytosis and Review of the Literature. Case Reports in Radiology.
Publisher | Google Scholor - Gallo JR, Paira S, Hernandez-Molina G, Delgado de la Mora J, Montante-Montes de Oca D. et.al. Immunoglobulin G4-associated Rosai-Dorfman Disease: Report of 3 Cases.
Publisher | Google Scholor - Foucar E, Rosai J, Dorfman R. (1990). Sinus histiocytosis with massive lymphadenopathy (Rosai -Dorfman disease): review of the entity. Semin Diagn Pathol, 7(1):19-73.
Publisher | Google Scholor - Faynea R, Sanchez Rengifoc S, Gonzalezb I, Solorzanoc JL, Gonzalez D. et.al. (2020). Primary cutaneous Rosai-Dorfman disease; a case-based review of a diagnostically and therapeutically challenging rare variant. Annals of Diagnostic Pathology, 45:151446.
Publisher | Google Scholor












