

Case Report
Undetectable Intrinsic Coagulation Factors in A Patient with Lupus Anticoagulant-Hypoprothrombinemia Syndrome
1 UniUPO, Department of Clinical Pathology and Clinical Biochemistry, Novara, Italy.
2 UniTo, Department of Clinical Pathology and Clinical Biochemistry, Torino, Italy.
3 SC Laboratory Analysis, AO Ordine Mauriziano, Torino, Italy.
*Corresponding Author: Hoza Roxana Maria, UniUPO, Department of Clinical Pathology and Clinical Biochemistry, Novara, Italy.
Citation: Hoza R. Maria, Maria P., Alessandra R., Domenico C., Barbara M. (2025). Undetectable Intrinsic Coagulation Factors in A Patient with Lupus Anticoagulant-Hypoprothrombinemia Syndrome. Journal of Hematology Research and Blood Disorders, BioRes Scientia Publishers. 2(1):1-5. DOI: 10.59657/jhrbd.brs.25.008
Copyright: © 2025 Hoza Roxana Maria, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: September 09, 2025 | Accepted: September 23, 2025 | Published: October 06, 2025
Abstract
Background/Objectives: Lupus anticoagulant–hypoprothrombinemia syndrome (LA-HPS) is a rare autoimmune disorder characterized by the coexistence of lupus anticoagulant (LA) and acquired hypoprothrombinemia due to anti–factor II antibodies. Patients face a paradoxical risk of both bleeding and thrombosis, representing a major diagnostic and therapeutic challenge.
Methods: We report the case of a 42-year-old male with a history of unprovoked venous thrombosis 12 years earlier. He was treated with oral anticoagulation for six months and, due to repeatedly equivocal lupus anticoagulant results (positive/negative in different laboratories), he was subsequently maintained on aspirin. He was referred to our laboratory for evaluation of prolonged PT and aPTT. Mixing studies, coagulation factor assays, inhibitor testing, ELISA, and thrombin generation assays were performed to identify the underlying abnormality.
Results: Mixing studies suggested the presence of an inhibitor. Initial factor assays using a silica-based aPTT reagent falsely indicated deficiencies of factors IX, XI, and XII. Repeat testing with an ellagic acid–based reagent normalized these factors, confirming LA interference. In contrast, factor II activity remained markedly reduced (15%), consistent with hypoprothrombinemia. ELISA demonstrated high titers of anti–phosphatidylserine/prothrombin, anticardiolipin, and anti–β2 glycoprotein I antibodies, establishing the diagnosis of LA-HPS. Thrombin generation testing revealed reduced peak height and endogenous thrombin potential, consistent with a hypocoagulable state despite the absence of clinical bleeding or thrombotic events. The patient was managed conservatively with close monitoring and no immunosuppressive therapy.
Conclusion: This case highlights the diagnostic complexity of LA-HPS and the critical role of reagent selection in coagulation testing, as LA can mimic multiple factor deficiencies. LA-HPS should be suspected in patients with prolonged PT and aPTT with strong antiphospholipid antibody positivity. Careful monitoring is essential to balance bleeding and thrombotic risks.
Keywords: lupus anticoagulant–hypoprothrombinemia syndrome; lupus anticoagulant; hypoprothrombinemia; antiphospholipid antibodies; coagulation testing
Introduction
Antiphospholipid syndrome (APS) is an autoimmune disorder characterized by thrombosis and/or obstetric complications in association with persistently positive antiphospholipid antibodies (aPL): lupus anticoagulant (LA), IgG and/or IgM anticardiolipin (aCL) and/or anti-β2-glycoprotein I (a-β2GPI) [1,2]. However, LA can also cause bleeding complications, which may be severe. Bajaj et al. first described a hemorrhagic disorder characterized by the concomitant presence of LA and hypoprothrombinemia caused by autoantibodies against factor II (prothrombin), later defined as Lupus Anticoagulant-Hypoprothrombinemia Syndrome (LA-HPS) [3]. Only a few cases have been reported in the literature, and about 50% of patients present with bleeding, ranging from minor episodes such as epistaxis to severe events such as gastrointestinal, muscular, or intracranial hemorrhage, particularly when factor II (FII) levels are very low (<10>
Materials and methods
This report was conducted according to ethical principles consistent with the Declaration of Helsinki. Since this article used only de-identified patient records and did not involve the collection, use, or transmission of individually identifiable data, it was exempt from Institutional Review Board approval. The medical literature was reviewed by searching both the MEDLINE and EMBASE databases.
Prothrombin Time (PT), activated Partial Thromboplastin Time (aPTT), Thrombin Time (TT), lupus anticoagulant (LA), and clotting factor activities were measured on an ACL TOP automated blood coagulation analyzer (Werfen, Bedford, MA, USA) using reagents provided by HemosIL Werfen (Bedford, MA, USA) and Siemens (Munich, Germany). aCL and a-β2GPI antibodies were measured on a Phadia automated analyzer (ThermoFisher Scientific, Waltham, MA, USA). aPS/PT antibodies were tested by ELISA (HemosIL Werfen, Bedford, MA, USA). Thrombin Generation Testing was performed using the BleedScreen assay on the ST Genesia automated analyzer (Stago, Asnières-sur-Seine, France).
Case presentation
In January 2025, a 42-year-old male outpatient presented to our laboratory for coagulation factor screening. He had a history of lower limb venous thrombosis, which had been treated with vitamin K antagonists (VKAs) for six months. Due to repeatedly equivocal lupus anticoagulant results (alternating positive and negative across different laboratories), he was subsequently maintained on aspirin.
First-level coagulation assays revealed a normal TT (ratio = 1.01; reference range: 0.75–1.20), prolonged PT (ratio = 1.63; reference range: 0.80–1.21), and prolonged aPTT with two different reagents: silica-based aPTT ratio = 2.16 (SynthASil, IL Werfen; reference range: 0.80–1.21) and ellagic acid–based aPTT ratio = 1.51 (Actin FS, Siemens; reference range: 0.70–1.18).
To differentiate between factor deficiencies and inhibitors against specific clotting factors or LA interference, PT and aPTT were repeated on a plasma sample mixed with normal plasma. In this assay, lack of correction suggests the presence of an inhibitor (such as LA or factor inhibitors), while correction indicates a coagulation factor deficiency. In our patient, both PT and aPTT mixing tests failed to correct [PT mixing ratio = 1.24 (normal range: 0.80–1.21); aPTT baseline mixing ratio = 1.85 (normal range: 0.80–1.18); aPTT mixing test after 2 h at 37 °C =1.91 (normal range: 0.80–1.18)], strongly suggesting the presence of an inhibitor.
Based on the general practitioner’s request, we assessed the activity of coagulation factors II, V, VII, VIII, IX, X, XI, and XII. Intrinsic factor assays were performed using a silica-based aPTT reagent (SynthASil, Werfen) on the ACL TOP analyzer, which is known to be sensitive to both factor deficiencies and lupus anticoagulant (LA). Results showed undetectable levels of intrinsic coagulation factors IX, XI, and XII and normal factor VIII levels. Common and extrinsic coagulation factors were measured using a PT recombinant thromboplastin (ReadiPlastin, Werfen); all factors were within the normal range, with the only exception of factor II, which showed markedly reduced activity (factor II = 15%; normal range: 60.0 - 150.0) (see Table 1).
Coagulation Factors
Table 1: Laboratory characteristics of our patient with LA-HPS
| Coagulative method | ||
| Assay | Results | Normal Values |
| Factor II (%) | 15 | (60.0 - 150.0) |
| Factor V (%) | 65 | (60.0 - 150.0) |
| Factor VII (%) | 92.4 | (60.0 - 150.0) |
| Factor X (%) | 83.3 | (60.0 - 150.0) |
| Factor VIII (%) | 76 | (50.0 - 200.0) |
| Factor IX (%) | 6.7 | (50.0 - 140.0) |
| Factor XI (%) | 0.4 | (50.0 - 150.0) |
| Factor XII (%) | 0.6 | (50.0 - 200.0) |
Further testing revealed strong positivity for LA screening, mixing and confirmatory Silica Clotting Time (SCT) and Dilute Russel Viper Venom Time (DRVVT) assays and to IgG anticardiolipin (aCL) and anti-β2-glycoprotein I (aβ2GPI) immunoassays (see Table 2).
Lupus Anticoagulant (LA)
Table 2: Anti-phospholipid antibodies measurement
| Coagulative method | ||
| Assay | Results | Normal Range |
| aPTT LA Screening (SCT) (Ratio) | 3.99 | (less than 1.28) |
| aPTT Mix (Ratio) | 3.16 | (less than 1.17) |
| aPTT Confirm (LA Normalized Ratio) | 1.67 | (less than 1.19) |
| DRVVT Screening (Ratio) | 6.73 | (less than 1.24) |
| DRVVT Mix (Ratio) | 5 | (less than 1.16) |
| DRVVT Confirm (LA Normalized Ratio) | 3.31 | (less than 1.18) |
| In solid phase antibodies | ||
| CliA method | ||
| aCL IgM (MPL/mL) | 19.3 | (99th percentile less than 19) |
| aCL IgG (GPL/mL) | 5287 | (99th percentile less than 14) |
| a-β2GPI IgM (U/mL) | 24.8 | (99th percentile less than 12) |
| a-β2GPI IgG (U/mL) | 14170 | (99th percentile less than 18) |
aPTT, activated Partial Thromboplastin Time; SCT, Silica Clotting Time; DRVVT, Dilute Russell's Viper Venom Time; aCL, anticardiolipin antibodies; a-β2GPI, beta-2-glycoprotein I antibodies.
Considering the possible interference of LA with coagulation assays, we repeated factors IX, XI, and XII measurements using an aPTT reagent known to be minimally sensitive to LA but highly sensitive to factor deficiencies (Actin FSL, Siemens) [6]. Using this ellagic acid-based aPTT assay, all intrinsic coagulation factors were within the normal range: factor IX = 131 (normal: 50–140), factor XI = 82% (normal: 50–150), and factor XII = 162% (normal: 50–200).
In the context of factor II deficiency, and in accordance with CLSI guidelines, we performed factor II assays on three dilutions of the patient’s plasma to assess linearity and parallelism. Non-parallelism is typically associated with LA or pathological inhibitors, whereas parallelism indicates factor deficiency. The ACL TOP software allows parallelism to be assessed by coefficient of variation (CV). After correcting for dilution factors, results are considered acceptable (parallel) if the deviation from the mean is ≤15%. In our patient, the “parallelism” CV was 1.5%, confirming strong inhibitor interference even in PT-based factor assays. We performed Nijmegen-Bethesda assay to measure Factor II inhibitors that showed a titer of 1.75 Bethesda Unit (normal value <0>
Given the strong aPL profile and the low factor II level, we also investigated the presence of aPS/PT antibodies. ELISA confirmed high IgG and IgM aPS/PT antibody titers (>157 U/mL). Altogether, these findings classify our patient as affected by LA-HPS.
Finally, to evaluate the overall hemostatic capacity and to assess both bleeding and thrombotic risk, we performed a Thrombin Generation Test. Results showed a reduction in peak height (the maximum thrombin concentration generated), and a marked reduction in endogenous thrombin potential (the total thrombin generated over time) compared with reference plasma, and normal subjects suggesting a bleeding tendency in this patient (see Figure 1).
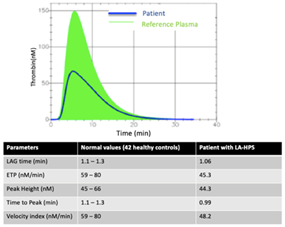
Figure 1: Thrombin Generation Curve
The bold curve represents the patient’s thrombogram, while the higher and thin curve represents the normal reference thrombogram. ETP = Endogenous Thrombin Potential.
Discussion and conclusions
Lupus anticoagulant–hypoprothrombinemia synd-rome (LA-HPS) is a rare autoimmune disorder characterized by the presence of lupus anticoagulant (LA) and acquired hypoprothrombinemia due to antibodies directed against factor II [6]. This syndrome is associated with an increased risk of bleeding, but paradoxically, several thrombotic events have also been reported [4,8–10]. LA-HPS should be suspected when both PT and aPTT are prolonged in the presence of LA positivity. Owing to its rarity, only a limited number of cases have been described in the literature.
There are currently no standardized guidelines for the treatment and management of LA-HPS. The mainstay of therapy generally consists of supportive measures (such as red cell transfusions, factor concentrates, and other blood components) combined with immunosuppressive therapy, including corticosteroids, cyclophosphamide, and other agents [11]. In some cases—particularly drug-induced or infection-associated LA-HPS—prothrombin deficiency may resolve spontaneously without the need for further treatment.
In this case report, we describe a 42-year-old male who was referred to our laboratory for the assessment of clotting factors II, V, VII, VIII, IX, X, XI, and XII, as requested by his general practitioner after first-level coagulation tests revealed prolonged PT and aPTT. The patient had history of unprovoked venous thrombosis 12 years earlier. He was treated with oral anticoagulation for six months and, due to repeatedly equivocal lupus anticoagulant results (positive/negative in different laboratories), he was subsequently maintained on aspirin.
Initial routine coagulation assays confirmed prolonged PT and aPTT, with no correction upon mixing studies, suggesting the presence of an inhibitor rather than a true factor deficiency. When factor assays were performed with a silica-based aPTT reagent (sensitive to aPL), factors IX, XI, and XII appeared undetectable. This finding underscores the importance of reagent selection in patients with suspected LA, since different activators vary in their susceptibility to aPL interference. When the assays were repeated with an ellagic acid–based reagent (Actin FSL), the intrinsic factor levels were within the normal range, confirming that the previous abnormalities were artifactual and due to aPL interference rather than genuine deficiencies.
In contrast, factor II deficiency persisted across assays and was further validated through parallelism testing. Together with markedly elevated aPS/PT antibody titers, these results established the diagnosis of LA-HPS. aPS/PT antibodies, which target prothrombin without neutralizing its coagulant activity, result in reduced circulating prothrombin antigen secondary to rapid clearance of antigen–antibody complexes by the reticuloendothelial system.
Despite the laboratory evidence of a bleeding tendency, the patient reported history of thrombosis. However, thrombin generation testing demonstrated reduced thrombin peak height and endogenous thrombin potential supporting the presence of a functional hypocoagulable state that could predispose the patient to bleeding if not closely monitored.
Our patient did not receive any immunosuppressive therapy to manage LA-HPS.
Given the potential risk of bleeding, careful clinical monitoring is warranted, particularly if anticoagulation becomes necessary for future thrombotic events. Moreover, the strong aPL profile—characterized by triple positivity and high titers of aCL, a-β2GPI, and aPS/PT antibodies—together with the patient’s history of venous thrombosis, suggests a high-risk APS phenotype, underscoring the need for ongoing clinical vigilance.
LA-HPS should be suspected in patients presenting with prolonged PT and/or aPTT. The management of PT and factor II recovery in LA-HPS requires careful balancing of bleeding and thrombotic risks. This case highlights the diagnostic complexity posed by APS-related interferences in coagulation testing. Accurate interpretation of coagulation assays in the presence of aPL requires caution, as reagent sensitivity can otherwise lead to misdiagnosis of factor deficiencies.
References
- Qiao, J., Bailly, J., & Opie, J. (2024). Key issues at the forefront of diagnosis and testing for antiphospholipid syndrome. Clinical and Applied Thrombosis/Hemostasis, 30:10760296241306751.
Publisher | Google Scholor - Devreese, K. M. J., Bertolaccini, M. L., Branch, D. W., de Laat, B., Erkan, D., Favaloro, E. J., et al. (2025). An update on laboratory detection and interpretation of antiphospholipid antibodies for diagnosis of antiphospholipid syndrome: Guidance from the ISTH-SSC Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibodies. Journal of Thrombosis and Haemostasis, 23(2):731–744.
Publisher | Google Scholor - Bajaj, S. P., Rapaport, S. I., Fierer, D. S., Herbst, K. D., & Schwartz, D. B. (1983). A mechanism for the hypoprothrombinemia of the acquired hypoprothrombinemia-lupus anticoagulant syndrome. Blood, 61(4):684–692.
Publisher | Google Scholor - Mulliez, S. M., De Keyser, F., Verbist, C., Vantilborgh, A., Wijns, W., Beukinga, I., et al. (2015). Lupus anticoagulant-hypoprothrombinemia syndrome: Report of two cases and review of the literature. Lupus, 24(7):736–745.
Publisher | Google Scholor - Gago, L., Lourenço, M. H., Torres, R. P., Mourão, A. F., Costa, M. M., Branco, J. C., et al. (2024). Lupus anticoagulant-hypoprothrombinemia syndrome: A duality between thrombosis and hemorrhage. Cureus, 16(11):e73149.
Publisher | Google Scholor - Kumano, O., Ieko, M., Naito, S., Yoshida, M., & Takahashi, N. (2012). APTT reagent with ellagic acid as activator shows adequate lupus anticoagulant sensitivity in comparison to silica-based reagent. Journal of Thrombosis and Haemostasis, 10(11):2338–2343.
Publisher | Google Scholor - Fujiwara, K., Shimizu, J., Tsukahara, H., & Shimada, A. (2019). Lupus anticoagulant-hypoprothrombinemia syndrome and immunoglobulin-A vasculitis: A report of Japanese sibling cases and review of the literature. Rheumatology International, 39(10):1811–1819.
Publisher | Google Scholor - Bel Feki, N., Zayet, S., Ben Ghorbel, I., & Houman, M. H. (2016). Lupus anticoagulant-hypoprothrombinemia syndrome presenting with co-existing cerebral venous thrombosis and subdural hemorrhage. Journal des Maladies Vasculaires, 41(6):403–406.
Publisher | Google Scholor - Mazodier, K., Arnaud, L., Mathian, A., Costedoat-Chalumeau, N., Haroche, J., Frances, C., et al. (2012). Lupus anticoagulant-hypoprothrombinemia syndrome: Report of 8 cases and review of the literature. Medicine (Baltimore), 91(5):251–260.
Publisher | Google Scholor - Chen, X., Nedved, D., Plapp, F. V., & Cunningham, M. T. (2018). Fatal pulmonary embolism and pulmonary hemorrhage in lupus anticoagulant hypoprothrombinemia syndrome: A case report and review of literature. Blood Coagulation & Fibrinolysis, 29(8):708–713.
Publisher | Google Scholor - Pilania, R. K., Suri, D., Jindal, A. K., Kumar, N., Sharma, A., Sharma, P., et al. (2018). Lupus anticoagulant hypoprothrombinemia syndrome associated with systemic lupus erythematosus in children: Report of two cases and systematic review of the literature. Rheumatology International, 38(10):1933–1940.
Publisher | Google Scholor























